
- 1one piece_娜美_01_one piece hentai
- 2CrossOver软件2023破解激活码_crossover-wine注册
- 3Spring Boot(四):Thymeleaf 使用详解_spring thymeleaf
- 4NX+Ubuntu18.04+ROS Realsense(RealSenseD435i )的安装与使用_realsense安装
- 5记录:rosdep update
- 6js通过a标签的方式下载文件并对其重命名的完整方案_a标签下载文件重命名
- 7JavaScript中的Array.prototype.forEach()方法(简介+重写)_js array.prototype.foreach
- 8leetcode 94 二叉树的中序遍历(java)_lecode中树的输入root = [1,null,3,2,4,null,5,6]是怎么转化成节点的
- 9R语言入门笔记2.1
- 10c#--正则表达式(项目常用)_c# 正则表达式 数字
【论文阅读】深度学习与多种机器学习方法在不同的药物发现数据集进行对比_深度学习论文对比实验
赞
踩
机器学习方法在医药研究中已经应用了几十年。与贝叶斯方法相结合的指纹类型分子描述符的相对易用性和可用性使得该方法广泛应用于与药物发现相关的各种端点阵列。深度学习是最新的机器学习算法,从对接到虚拟筛选等许多制药应用都受到关注。
深度学习是一种基于多层隐层的人工神经网络,在许多人工智能应用中得到了广泛的应用。我们之前已经提出,需要将不同的机器学习方法与适用于药物研究的不同数据集阵列中的深度学习进行比较。与药物研究相关的终点包括吸收、分布、代谢、排泄和毒性(ADME/Tox)特性,以及对病原体的活性和药物发现数据集。
在本研究中,我们使用了溶解度、似度、hERG、KCNQ1、黑死病、恰加斯病、肺结核、疟疾等数据集,比较了使用FCFP6指纹的不同机器学习方法。这些数据集表示整个细胞屏幕、单个蛋白质、物理化学特性以及具有复杂端点的数据集。我们的目的是评估在使用AUC、F1分数、Cohen’s kappa、Matthews相关系数等一系列指标进行评估时,深度学习是否对测试有任何改善。
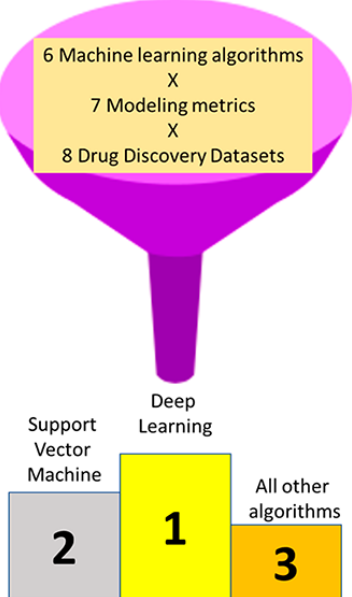
深度神经网络(Deep Neural Networks, DNN)对指标或数据集的归一化得分排序高于支持向量机(SVM),而SVM又高于其他所有机器学习方法。 使用雷达类型图可视化训练和测试集的这些属性,表明模型在什么时候是较差的或者可能是训练过度的。这些结果还表明,需要使用更大规模的比较、前瞻性测试以及不同指纹和DNN架构的评估等多个指标来进一步评估深度学习。
一、简介
药物发现目前正处于这样一个阶段:PubChem、ChEMBL以及越来越多由高通量筛选和高通量生物学(包括全细胞表型筛选、酶、受体等)创建的其他数据库的公共数据量不断增加,使其完全处于“大数据”领域。我们面临着重大的挑战。我们不再局限于少数分子及其性质,我们现在有成千上万的分子和几十个性质要考虑。我们如何挖掘、使用这些数据,并希望从中学习,从而使药物发现更有效、更成功?
一个方法是利用化学信息使用机器学习处理这些大数据的方法,如使用支持向量机(SVM), K近邻(KNN),朴素贝叶斯,决策树等已越来越多地使用。这些方法可以用于二进制分类、多类分类,或值的预测。
近年来,深度人工神经网络(包括卷积网络和递归网络)在模式识别和机器学习领域赢得了众多的竞争。深度学习通过引入以其他更简单的表示形式表示的表示来解决表示学习中的核心问题。n层神经网络如图1所示。
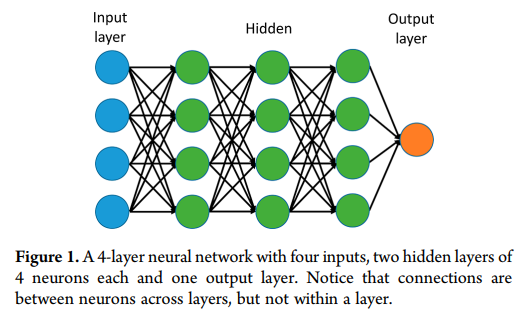
值得注意的是,单层神经网络描述的是一个没有隐含层的网络,其中输入直接映射到输出层。在这个意义上,逻辑回归或支持向量机只是单层神经网络的一个特例。在我们的工作中,为了简化深度神经网络(DNN)的表示,我们将只计算隐藏层。通常1−2隐藏层神经网络被称为浅神经网络和3或更多的隐藏层神经网络被称为深层神经网络。
最近的一篇综述讨论了深度学习在药物研究中的发展和应用,这种方法在其他地方的图像和语言学习中被证明是非常成功的。以前深度学习主要用于无监督学习和噪声数据。将深度学习用于药物应用的有限努力表明,与其他方法相比,需要进一步探索其在化学信息学方面的效用。
深度学习在生物信息学和计算生物学中得到了较为广泛的应用。深度学习也被用于预测性质,如水溶性,使用四个公布的数据集,并与其他机器学习方法比较显示出良好的10倍交叉验证(10-fold cross validation)结果。
到目前为止,Merck已经对深层神经网络进行了比较,并将其与随机森林进行了比较,以使用大型定量结构活动关系(QSAR)数据集。他们发现在15个数据集中的11个表现要好于随机森林,在第二次使用时间分割测试集的评估中,15个数据集中的13个表现优于随机森林。但是Merck没有研究其他的机器学习方法。与其他机器学习方法一样,深度学习模型得到验证的最大例子之一是Tox21挑战。在核受体和应激反应数据集上,多任务学习的深度学习略优于最接近共识的ANN方法。最近,有一个小组提出了一些分子机器学习的数据集,并将这些数据集与选定的机器学习方法进行了比较。第二组用7个ChEMBL数据集评价了若干机器学习方法,但只着重于评价性能的单一指标。深度学习常常单独应用于单个数据集,而不是与许多可用的替代方法进行比较。有可能有更多的数据集可以从深度学习中受益,尽管它们可能更小。
这些机器学习方法越来越多地用于化合物的虚拟筛选,通过用活性化合物丰富筛选的化合物集,可以更有效地利用高通量筛选(,HTS)资源。此外,这种机器学习方法还可以用于药物的吸收、分布、代谢、排泄和毒性(ADME/Tox)特性,因为这些因素可以影响药物发现过程的成功,并且它们的早期评估可以预防失败。过去的研究表明这些计算方法可以极大地影响药物发现效率。
在过去的十年中,我们和其他人越来越关注贝叶斯方法,因为它们的易用性和一般适用性,使用最大直径6的分子函数类指纹和其他几个简单的描述符。这项工作的大部分集中在考虑细胞毒性的结核分枝杆菌模型上,并对其进行前瞻性评估,以显示与随机筛查相比,结核分枝杆菌的高命中率。此后,我们利用查加斯病和埃博拉病毒的数据集,对批准的药物以及模型ADME特性(如水溶性、小鼠肝微粒体稳定性、Caco-2细胞通透性、62个毒理学数据集和转运体)进行了重新利用。通过制作指纹,以及贝叶斯模型构建算法的开源,有潜力进一步拓展这方面的工作。
本研究的主要目的是 评估在药物发现和ADME/Tox数据集的其他计算方法中,使用一系列指标进行评估时,深度学习是否对测试有任何改善。在此过程中,我们开发了一种方法,使深度学习模型更容易获取。
二、实验
2.1 实验环境
所有的计算都是在一个双核处理器,四核(英特尔E5640)服务器上完成的,运行CentOS 7, 96GB内存和两个特斯拉K20c GPU。
安装了以下软件模块:
nltk 3.2.2、scikit-learn 0.18.1、Python 3.5.2、Anaconda 4.2.0(64位)、Keras 1.2.1、Tensorflow 0.12.1、Jupyter Notebook 4.3.1。
2.2数据集和描述符
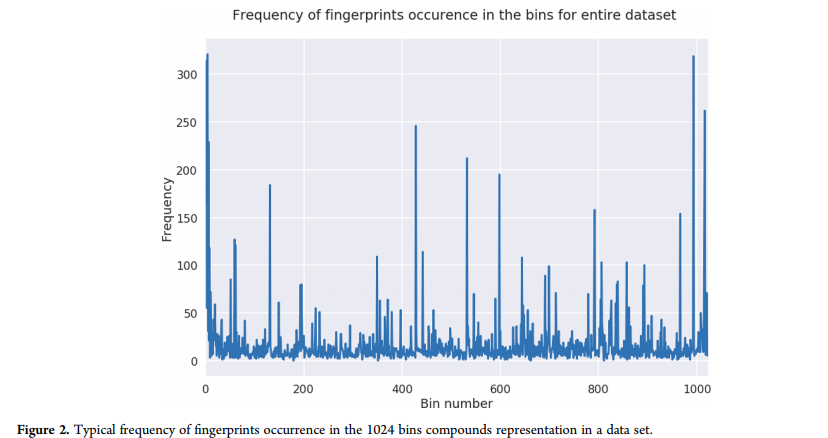
利用公开的用于不同类型活性预测的不同药物发现数据集开发预测管道(表1)。Clark等人使用相同的数据集,探索一系列贝叶斯模型在ADME/Tox等理化性质预测中的适用性。在目前的FCFP6指纹研究中,使用RDKit (http://www.rdkit.org/)从SDF文件中计算出1024个bin数据集。在图2所示的数据集中,指纹出现的典型频率出现在1024个容器的复合表示中。

2.3机器学习
开发了两个通用预测方法。
第一个方法仅使用经典机器学习(CML)方法构建,如伯努利朴素贝叶斯、线性逻辑回归、AdaBoost决策树、随机森林和支持向量机。开源的Scikit-learn (http://scikit-learn.org/stable/, CPU用于训练和预测)使用ML python库构建、调优和验证这个方法中包含的所有CML模型。
第二个方法使用不同复杂性的深度神经网络(DNN)学习模型构建,使用Keras (https://keras.io/)、一个深度学习库和Tensorflow (www.tensorflow.org, GPU training and CPU for prediction)作为后端。开发的方法包括将输入数据集随机分割为训练(80%)和测试(20%)数据集,同时在每次分割(分层分割)中保持活动类比与非活动类比的相等比例。因此,所有模型的调优和超参数搜索都是通过对训练数据进行4倍交叉验证来进行的,这样可以更好的进行模型泛化。提供了一个示例 Jupyter notebook。
【示例程序】
2.4 数据分析
使用到的评估方法:
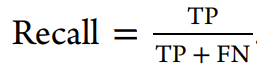
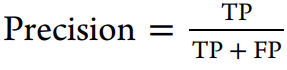
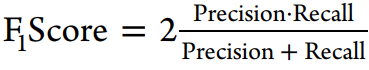
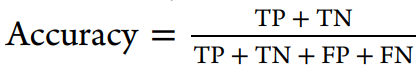
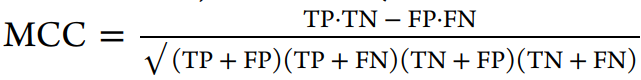
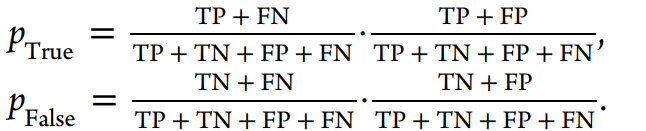
三、结果
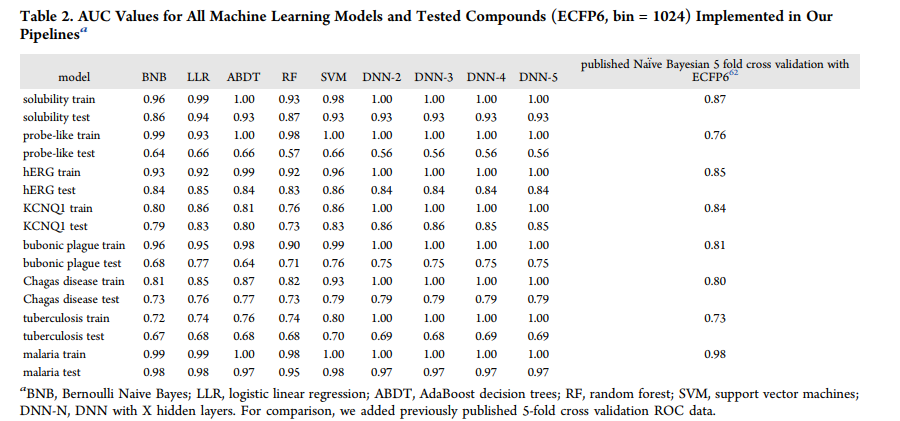
F1-score AUC,准确性,卡帕,马修斯相关性,精度和召回的所有训练值表示为模型化合物FCFP6指纹在1024箱总结(表2和表S1−16)虽然个别模型文件也提供(支持信息)。
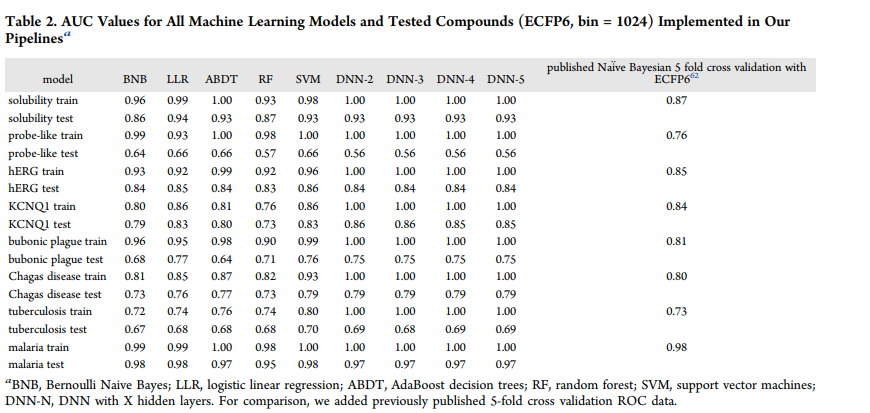
为了清晰起见,我们按照每个数据集、训练集和测试集对所有指标进行了分组,并将它们表示为雷达图。然而,在本例中,我们使用了不同的描述符和建模算法源FCFP6 vs ECFP6,以及RDKit vs CDK。在许多情况下,SVM模型的测试集通常比同一方法中的任何其他模型都要好。
当分析雷达图时,很容易看出哪些模型可能训练过度。在这种情况下,模型训练集的所有指标的得分都很高,而测试集的得分则低得多。图的形状也可以表示模型的质量。测试集的圆圈越大,模型就越好。
溶解度模型(图3)是一个很好的平衡模型的例子。训练集和测试集实际上都由相似的圆形图表示,很明显,BNB方法在大多数指标中表现最差。
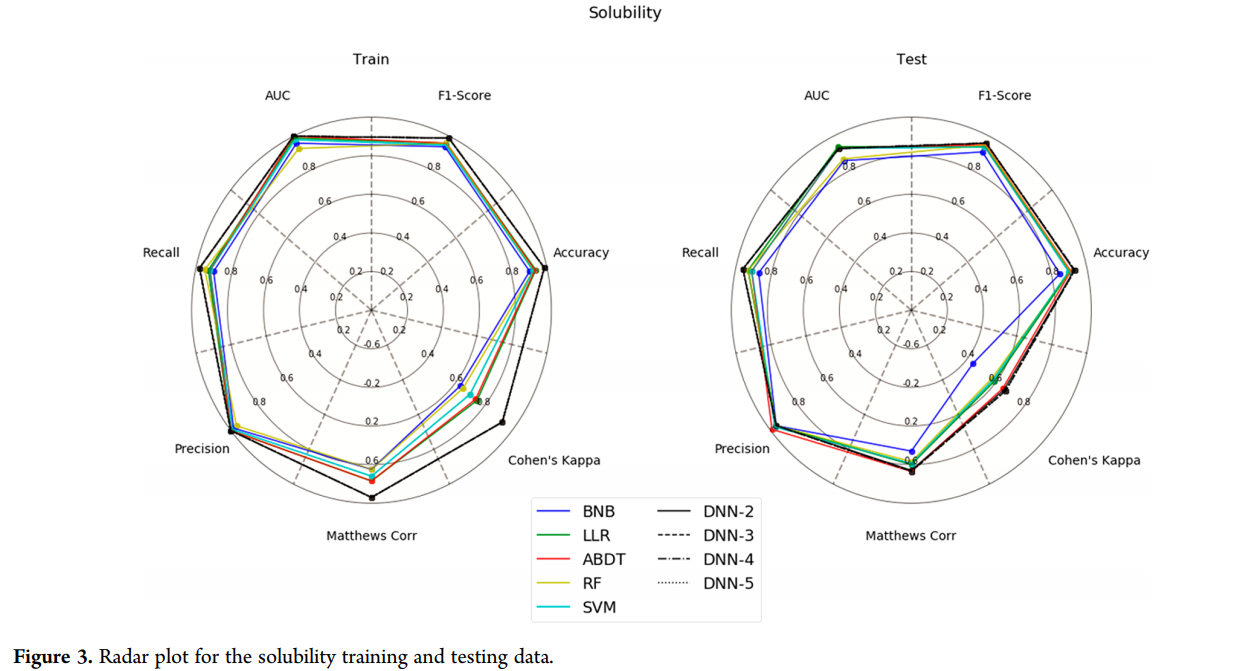
probe样模型(图4)对测试集的分数进行了不规则的排列,这表明它在所有方法中对Cohen 's Kappa的表现都很差。
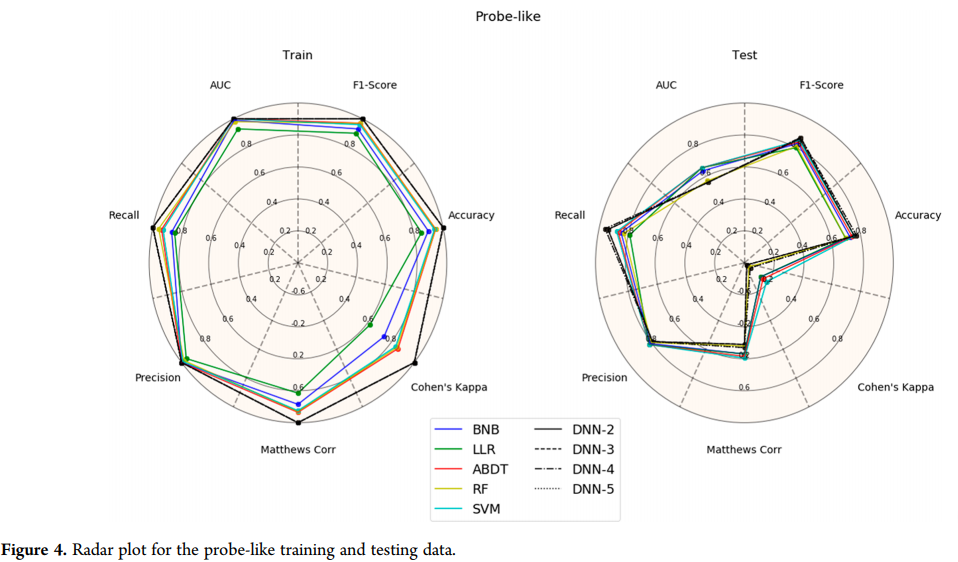
hERG模型的测试集(图5)显示,大多数方法在指标之间是可比较的,ABDT在测试集中表现最差
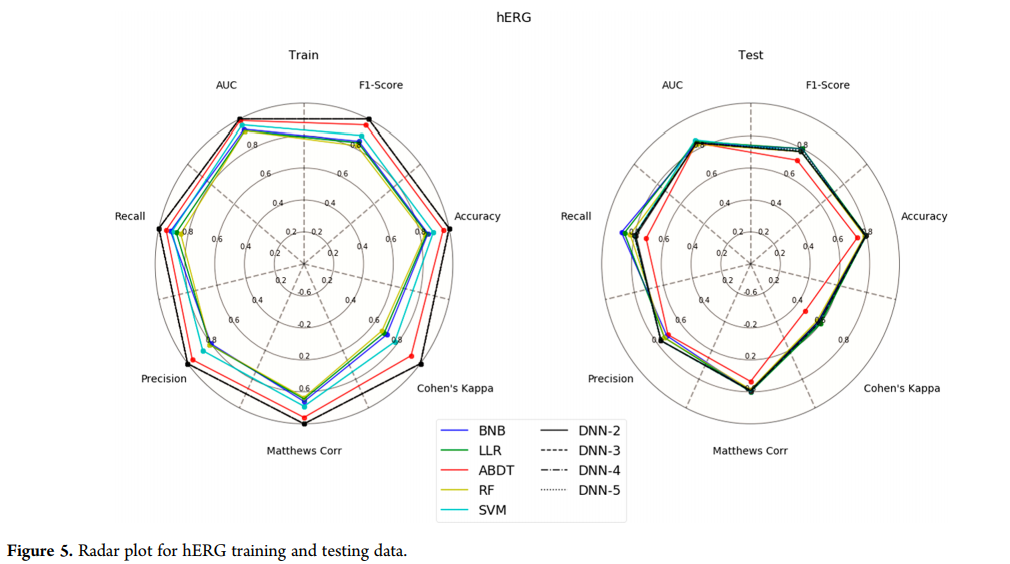
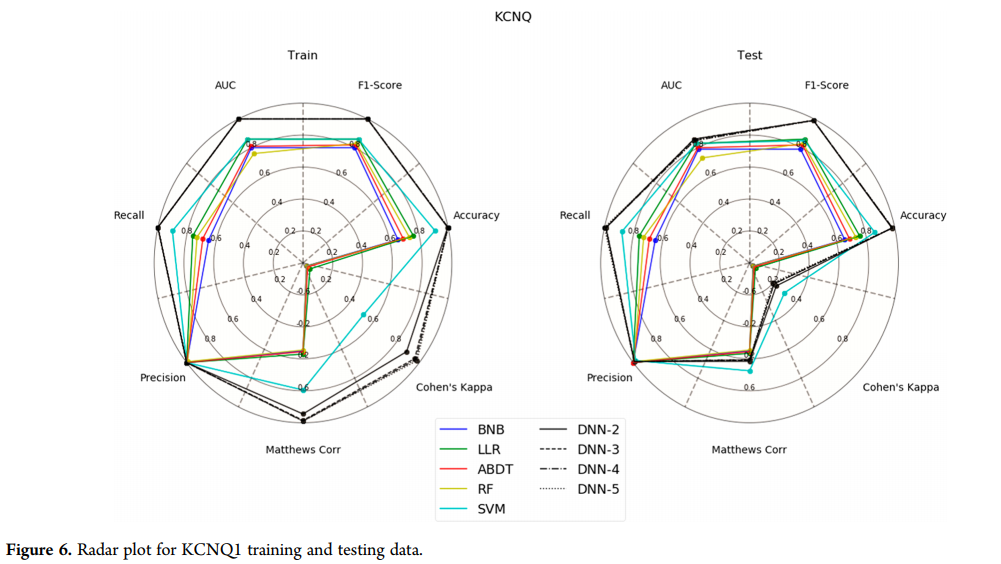
总体而言,Cohen’s Kappa是该数据集最敏感的度量标准。KCNQ1模型(图6)显示,DNN和SVM在训练和测试方面优于其他方法,Matthews相关性和Cohen’s Kappa评分显著低于所有其他指标
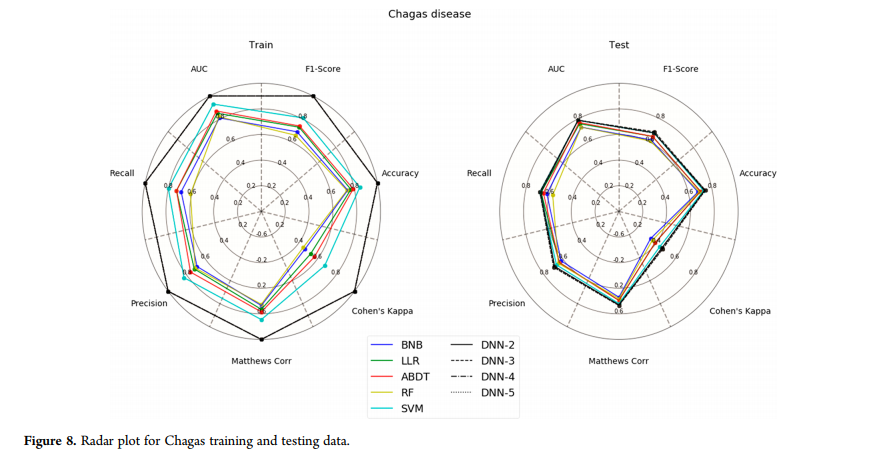
黑死病模型(图7)是一个困难的例子,DNN在训练和测试中很容易胜过所有方法(AUC, Matthews correlation and accuracy metric表现最好)。Chagas疾病数据集(图8)再次显示,以Cohen’s kappa为最敏感的指标,DNN具有较好的训练和测试性能。
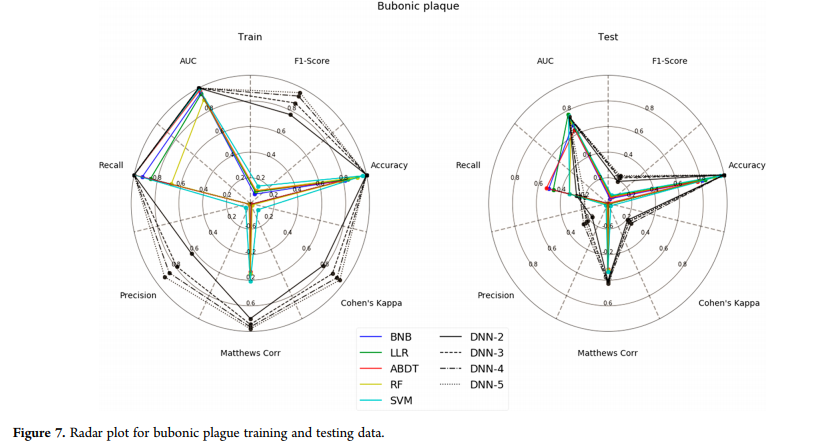
Chagas疾病数据集(图8)再次显示,以Cohen’s kappa为最敏感的指标,DNN具有较好的训练和测试性能
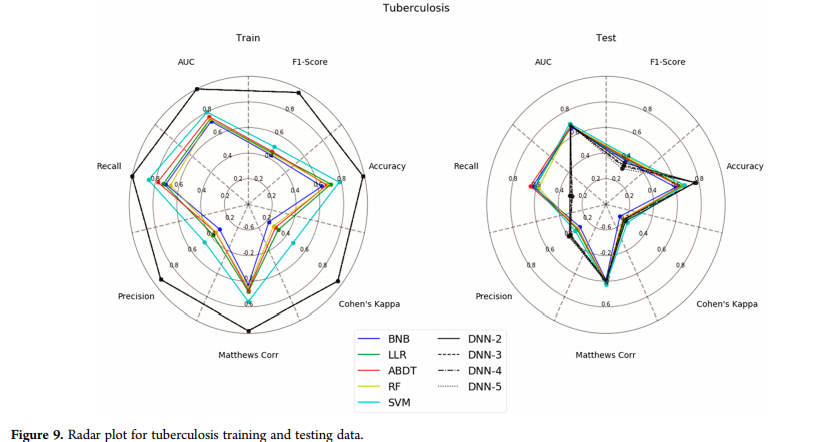
结核病数据集(图9)是另一个例子,除了召回统计数据外,DNN在培训方面比所有方法都要好得多,在测试集方面也比所有方法差得多。在测试集的所有方法中,精确度、f1评分和Cohen 's Kappa都很差。
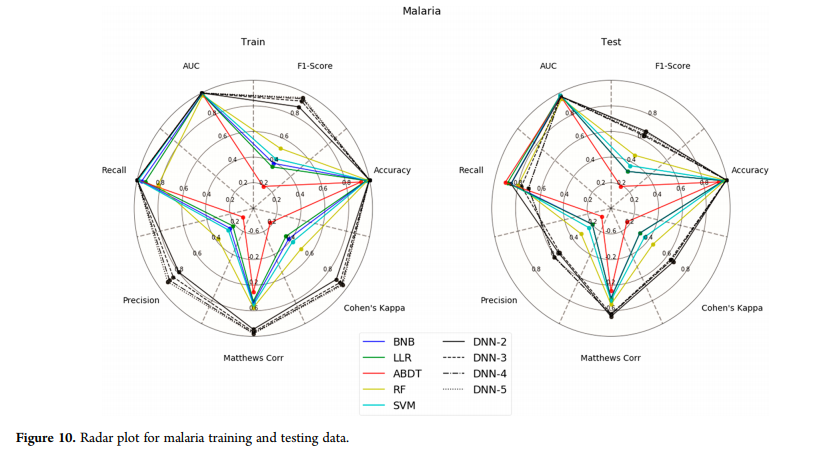
疟疾数据集(图10)显示了DNN对训练和测试集的影响,与其他机器学习方法相比,DNN在准确率、f1评分和Cohen’s Kappa方面都有很大提高
总的来说,除了probe样数据集的AUC性能外,DNN模型在外部测试集预测中表现良好。对于AUC, DNN-3在8个数据集中的6个上优于BNB(表2)。
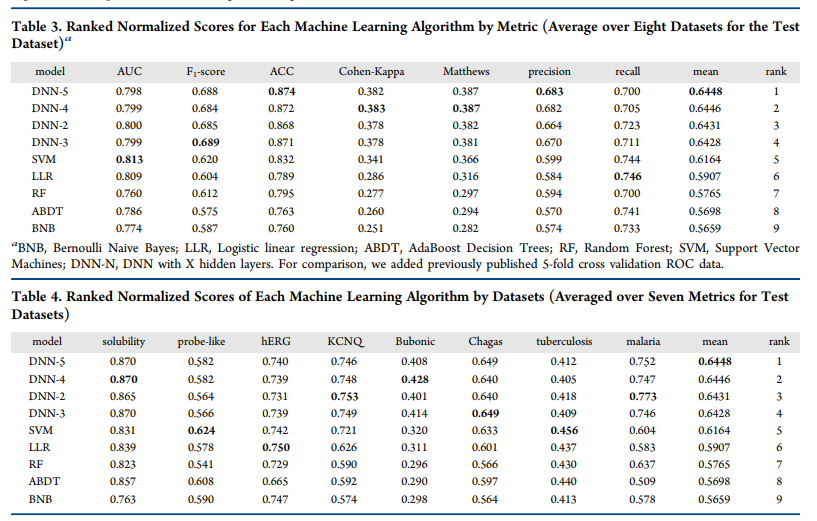
为了进一步了解哪种模型的性能最好,我们使用了按度量(表3)和数据集(表4)对每个机器学习算法进行排序的标准化得分。这种方法以前曾被其他人用于比较多种机器学习方法和性能标准。当模型以指标或数据集进行排序时,深度学习(DNN-5和DNN-4)的排名高于SVM,其他方法均低于此(表3和表4)。
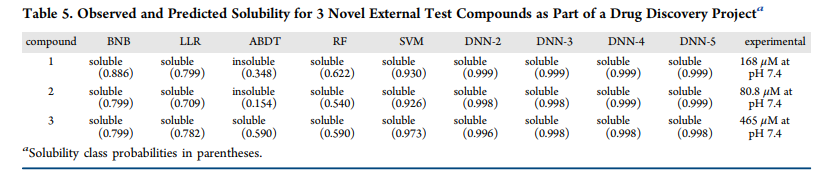
我们使用本研究中开发的所有不同的溶解度机器学习模型,对我们的一个药物发现项目中的3种化合物的溶解度进行了评估,如表5:
四、讨论
到目前为止,很少有研究将深度学习与广泛的经典机器学习方法进行比较,或者讨论了这种方法在药物研究中的应用,甚至使用模型对正在进行的项目进行实际预测。因此,本研究填补了这些方法在药物发现应用方面的空白。对于我们可能需要考虑的所有计算建模方法,它们都依赖于模型的适用性域,并受到底层数据质量的影响,而这些数据反过来又决定了任何模型和预测的实用性和相关性。将深度学习与其他机器学习算法在一系列应用中的比较表明,当主要使用内部交叉验证作为评估形式时,这种方法常常会得到改进。在本研究中,我们在所有情况下使用外部测试集来比较不同的算法。此外,我们还比较了几个用于评估绩效的指标,这与该领域大多数已发表的研究不同,后者依赖于一个指标或一个狭窄的范围。我们的比较方法使用了一种等级标准化的评分方法,这种方法已经被其他人用来比较机器学习算法和其他领域的性能指标。
本研究的结果显示,由于在训练和测试集评估的所有指标上存在差异,要全面改进深度学习模型还有很多工作要做。另外,很明显,一些指标没有其他指标那么敏感。例如,AUC远没有Cohen Kappa那么敏感。因此,这可能是一个比AUC更有用的测试集度量。然而,单独使用AUC我们可以辨别不同的模型与外部测试(表2)。但是,单独使用AUC,我们可以看出模型与外部测试的差异(表2),8个数据集中至少有7个数据集从贝叶斯改进到支持向量机,8个数据集中有5个数据集从贝叶斯改进到DNN-3。这代表了真实的外部测试集的结果。在8种情况下,训练ROC从贝叶斯增加到DNN3。秩归一化得分法表明,DNN的性能优于SVM, SVM的性能又优于其他所有算法(表3和表4)。这些结果表明,SVM和DNN在药物发现的不同场景下,可能需要得到科学家的进一步接纳和更严格的评价。
撇开个别模型的缺点不谈,我们研究了其中一个模型的“现实世界”适用性,即溶解度,在最近的一个药物发现项目中。溶解度是一种物理化学性质,通常在药物开发项目的过程中,许多化合物的溶解度都是由这种性质决定的。 在这里,我们使用分类模型和回归模型来确定三个内部化合物的溶解度预测。8/9模型(包括所有款模型)正确识别所有三个化合物作为可溶性(溶解度> 10μM / L)。唯一的错误分类来自AdaBoost决策树模型,将两种不溶性较差的化合物标记为不溶性。值得注意的是,DNN的模型都将这些化合物标记为>99%的可溶性的概率。然后,我们使用线性分类器(弹性网络)和DNN模型评估了预测溶解度的回归性能。所有模型的精度都在约1个对数单位的解度范围内,并倾向于预测可溶度(表S7)无可否认,这是一个非常小的测试集,但它表示的化合物是高级先导物,因此具有相当大的意义。对这些溶解度机器学习模型的进一步前瞻性评估需要更多的分子,但这只是一个起点。
虽然我们在本研究中没有对不同的描述符进行评估,但是FCFP6在本研究的数据集中表现得很好 。然而,未来的研究可能会评估额外的描述符,例如其他具有深度学习的非指纹描述符。最近的一篇论文描述了分子图的卷积,它表示了一种更简单的分子编码,即机器学习应用中的无向原子图。利用不同的机器学习方法开发附加描述符及其评估,将有助于找到描述符和机器学习算法的最佳组合。我们没有观察到数据集大小或平衡有任何明显的影响,这可能是由于评估的数据集数量有限。
应该指出的是,直到最近,对于普通科学家来说,用于构建模型或生成预测的开放源码的化学信息学深度学习工具包还相对难以使用 。促进非专家用户使用机器学习将有助于增加这些方法对药物发现的潜在影响。理想情况下,这些和其他机器学习模型需要使用简单和透明。
由于我们之前对各种药物发现项目(使用商业软件构建)的贝叶斯模型的前瞻性测试已经为活体动物测试带来了一些有前途的线索和候选对象,模型统计数据的有意义的改进可能会提高前瞻性测试的命中率。我们还提出有必要将这些模型实际应用到真实的药物发现任务中,如评估溶解度(表5)。本研究的一个关键发现是,模型比较可能需要更广泛的模型度量,依赖AUC可能并不理想。在未来,我们可能还会使用这些和其他指标来评估深度学习在未来虚拟筛选中的表现,以及评估更大的数据集数组。这些案例研究正在进行中,对于理解深度学习和其他机器学习方法在药物发现方面的范围和局限性将是有价值的。

