
- 1【原创】如何利用分层测试概念设计针对性测试用例_分层测试用例设计
- 2从谷歌下载APK到电脑
- 3同个局域网内SSH远程Ubuntu系统_连接同一个网络的电脑如何ssh
- 4Android 快速开发框架:推荐10个框架:afinal、ThinkAndroid、andBase、KJFrameForAndroid、SmartAndroid、dhroid.._kjframeforandroid androidquick fastapp
- 5CPU、GPU、IPU、NPU、TPU、LPU、MCU、MPU、SOC、DSP、FPGA、ASIC、GPP、ECU、
- 6【初阶C语言】关于scanf函数的超详细介绍和多组输入
- 7MQTTBox下载及设置相关参数
- 8计算机结构体系:循环展开题型 (非凭感觉的方法详解)_daddiu
- 9python知识推理知识图谱_知识图谱系列--知识推理
- 10编程常用英语词汇
PNAS:密歇根大学张阳教授团队实现对任意人工蛋白构型的从头设计_replica exchange monte carlo
赞
踩
蛋白质是生命体执行功能的基本生物分子。蛋白质分子折叠结构的多样性赋予了它们独特而丰富的功能,从而也让生命世界丰富多彩。尽管理论上蛋白质序列和结构空间十分庞大,然而历经数十亿年进化后,只有极少部分蛋白空间出现于自然界。这可能是生命体在演化过程中为适应自然选择压力而做的最终选择。研究表明,目前的PDB数据库几乎涵盖了所有天然折叠,而SCOPe的蛋白拓扑构型数据库的统计显示这些天然折叠只含有不到1500种不同蛋白折叠类型。这些数据表明,蛋白质的理论结构空间和自然界展示的结构空间存在巨大差异。
为探索和弥补这种差异,也鉴于蛋白质对生命体的重要性,近年来利用计算机来从头设计具有比天然蛋白更强劲功能的人工蛋白质受到极大关注。然而,绝大部分尝试都是基于实验已经解析的天然蛋白结构来设计新的蛋白序列。虽然这些尝试能有效解决某些问题(比如功能优化),但是基于天然蛋白结构的设计存在很大局限性。因为大自然只进化并赋予了很有限的蛋白折叠类型,依赖于天然结构的蛋白设计只能在极为有限的结构空间内进行微调,从而大大制约它在实际研究(例如药物研发)中的应用。可以想象,所有天然蛋白折叠都无法胜任某项任务的极端情况也会存在。特别是,随着人工制造全新生命物质的可能性越来越高,这种需求会变得越来越普遍。因此,从头设计蛋白质,即不仅仅是基于已有天然结构来设计蛋白序列,而是设计全新的蛋白结构(例如新的折叠类型和药物结合口袋)和具有特定功能的蛋白序列,已经成为目前生命科学领域中一个重要前沿方向,受到了广泛关注!
针对蛋白构型从头设计这一重大基础性科学问题,美国密西根大学华人科学家张阳教授课题组开发了一款全新的蛋白构型设计工具:FoldDesign。该工具通过不依赖于蛋白序列(或称“无序列”)片段组装模拟,可以对任何蛋白质拓扑学构型进行高保真度、高稳定性的从头设计。该算法于近日发表于美国国家科学院院刊(PNAS),张阳教授为论文通讯作者,张阳实验室的博士研究生Robin Pearce、研究科学家黄小强博士分别为论文第一、第二作者(图1)。

图1. 《PNAS》在线发表论文
FoldDesign方法简述
图2描述了FoldDesign算法流程。FoldDesign以任意给定蛋白的二级拓扑结构序列作为输入,将其拆分为长度为1~20的片段,并从PDB结构库中搜索与这些片段具有相似二级结构的序列片段,并对检索到的片段结构进行模拟组装。因FoldDesign在设计蛋白结构时序列未知,作者开发了一套不依赖于蛋白序列的能量函数,在片段替换的基础上引入11种新的构象移动模式(如键长伸缩、键角弯曲、二面角旋转等),通过副本交换蒙特卡洛(replica-exchange Monte Carlo)模拟进行片段组装,生成大量保持初始二级结构约束的蛋白3D结构模型,并从中选择FoldDesign能量打分最佳构象进行原子水平的精调以产生最终蛋白结构模型。需要注意的是,FoldDesign并不设计蛋白序列。基于FoldDesign结构,蛋白序列设计工作可由张阳课题组先前开发的EvoDesign和EvoEF2或者其他蛋白序列设计软件来实现。
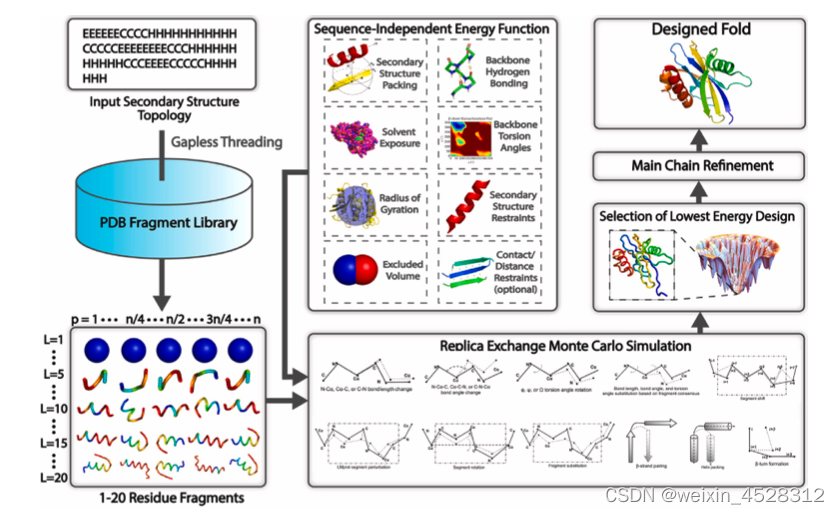
图2. FoldDesign算法流程图:利用无序列片段组装模拟实现全新蛋白质结构从头设计
实验结果及分析
需要指出的是,计算机自动产生一个稳定紧致、且满足人工需要的任意拓扑学二级结构的蛋白质构型并非易事。传统的、基于模板的蛋白构型算法,比如I-TASSER和Modeller,所构建的模型往往局限于已知的同源模板。即使加入新的约束限制,其最终模型仍难以偏离同源模板以产生有意义的、紧致的新结构。即使是从头预测算法,如Rosetta和QUARK,由于其序列依赖的能量项约束所限,其构造的模型往往聚焦于若干团簇,只能覆盖有限的折叠空间。
本研究中,为检验FoldDesign产生新蛋白构型的能力,作者构建了一个包含354个天然拓扑结构的数据集(涵盖α、β、α/β蛋白),然后对FoldDesign设计结构的能力进行系统性评估。作者首先考察了FoldDesign设计的蛋白折叠可保持(输入时)指定的二级结构序列的能力。为此,作者用DSSP获取354个蛋白的二级结构序列作为输入,通过FoldDesign设计3D结构后提取其二级结构序列并与输入的二级结构进行比较。作者采用Q3打分来量化设计的蛋白折叠对人工输入的二级结构的保真度,并对FoldDesign与Rosetta两个蛋白折叠设计方法进行比较。测试表明,FoldDesign折叠保持初始二级结构的能力显著优于Rosetta。FoldDesign在全数据集上Q3打分为87.7%,Rosetta为83.3%。FoldDesign在α蛋白上表现优异,Q3打分达到93.4%,而Rosetta只有82.8%。
尽管基于二级结构相似性的片段替换是FoldDesign组装3D折叠的基础,作者发现引入的新构象运动模式有助于生成能量打分更优的结构(图3A)并显著提高组装效率(图3B)。例如,只考虑片段组装时,蒙特卡洛模拟容易“卡”在特定的构象(图3C,水平线部分),而新的构象运动集可以很好地克服这种情况(图3D,不存在水平线)。这说明这些新的构象运动模式可以帮助FoldDesign探索更细致的构象空间,从而有利于生成能量更优的折叠。考虑新的构象运动集也让所设计的折叠更为紧凑(图3D),而只基于片段替换设计的折叠则较为松散(图3C)。这说明构象移动对基于片段组装的蛋白结构设计非常重要。
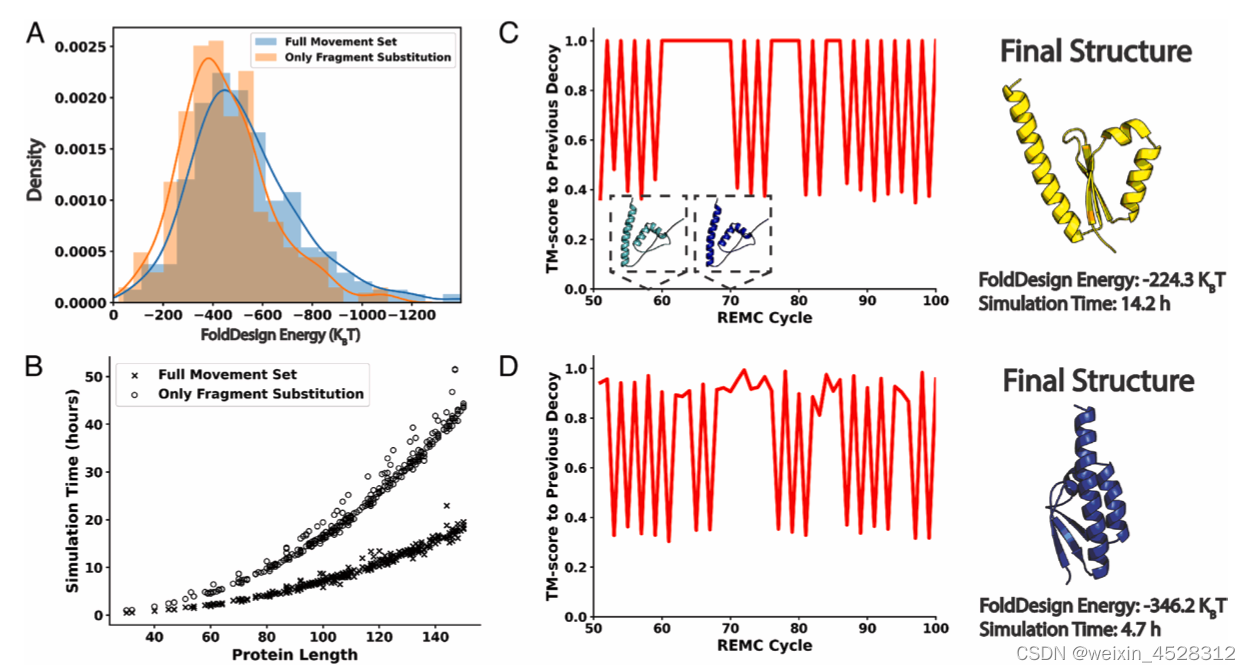
图3. 新的构象运动模式对FoldDesign从头设计至关重要
作者随后采用EvoEF2和RosettaFixBB对FoldDesign和Rosetta设计的354个结构进行蛋白序列从头设计。实验表明,无论用EvoEF2还是RosettaFixBB,在基于FoldDesign结构上设计的蛋白具有更高的残基包埋比例,并与天然蛋白的残基包埋比例相当(图4A)。相应地,基于FoldDesign结构设计的蛋白具有较小的溶剂可及表面,且与天然蛋白的溶剂可及表面相当(图4B)。作者采用两种高精度的蛋白结构评估统计势GOAP和ROTAS对设计的蛋白进行分析,发现基于FoldDesign的蛋白具有更优的GOAP/ROTAS能量打分(图4C-D)。这些结果表明,与其他的构型设计算法相比,FoldDesign设计的蛋白结构具有更好的包埋核心、更小的溶剂可及表面、以及更合理的三维拓扑(体现于更低的GOAP/ROTAS打分)。
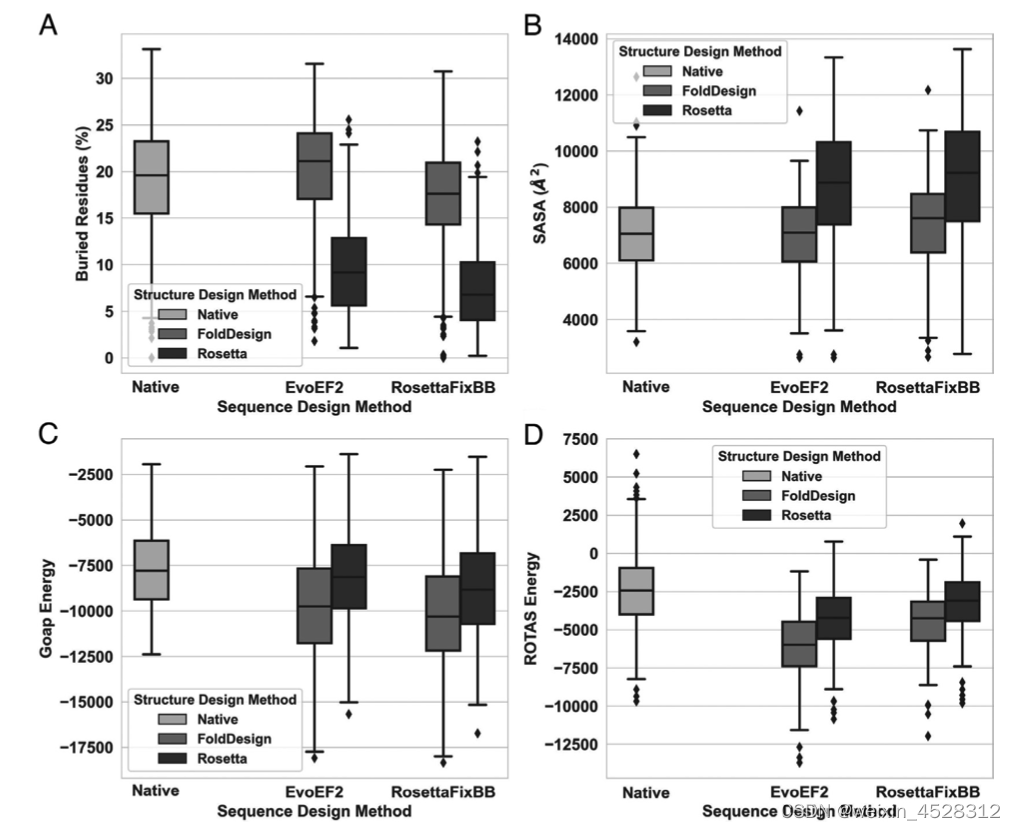
图4. FoldDesign与Rosetta设计的蛋白结构物理、能量等性质的对比
接下来,作者对EvoEF2/RosettaFixBB设计的序列能否折叠成FoldDesign生成的结构及其稳定性进行考察。作者采用两种方法进行评估:AlphaFold2结构预测和分子动力学模拟。总体上,基于FoldDesign结构所设计的序列由AlphaFold2预测的结构更接近初始FoldDesign结构(平均TM-score更高,图5C-D)。通过20纳秒分子动力学模拟表明,基于FoldDesign结构设计的蛋白能更好地保持初始FoldDesign结构(图5A-B)。这说明,与Rosetta相比,FoldDesign设计的构型更稳定。
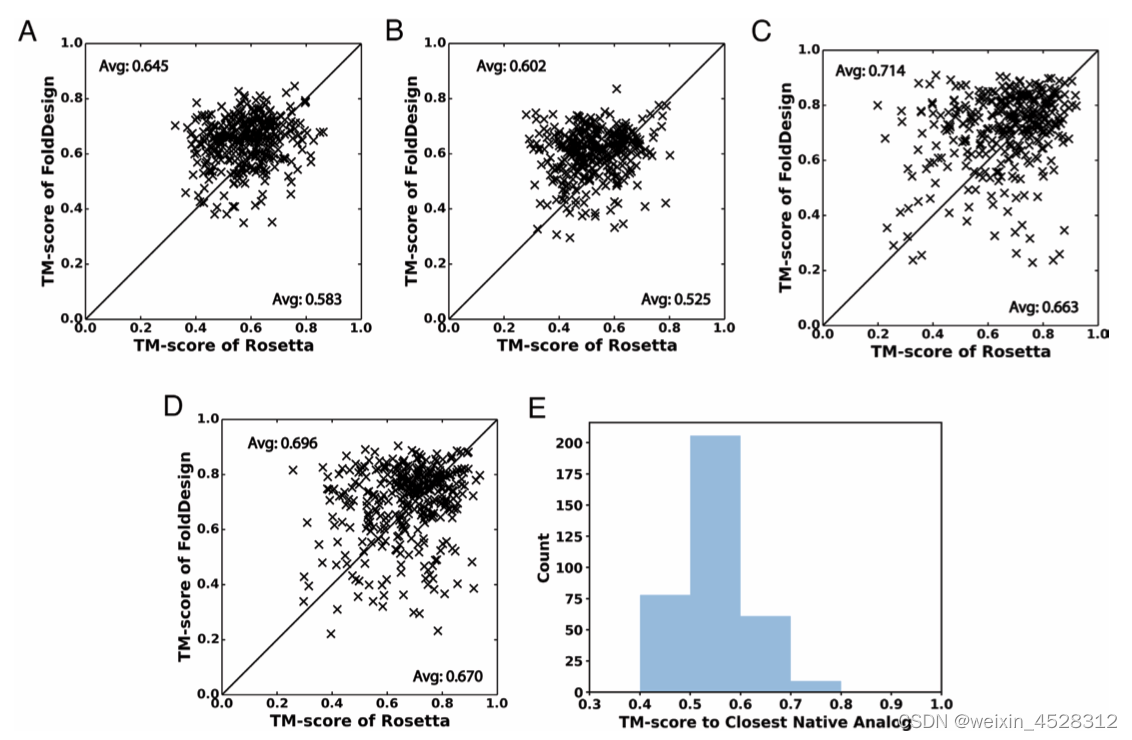
图5. 基于FoldDesign和Rosetta结构设计蛋白序列后的分子动力学模拟(A-B)及AlphaFold2结构预测分析(C-D)。图A/C基于EvoEF2设计序列,图B/D基于RosettaFixBB设计序列。(E)354个FoldDesign结构与PDB中最相似结构的TM-score分数的直方分布
作者进一步将最新的AI蛋白质结构预测软件AlphaFold2对其所设计的序列进行结构预测。图6直观地展示了FoldDesign设计的稳定的、折叠良好的蛋白结构。
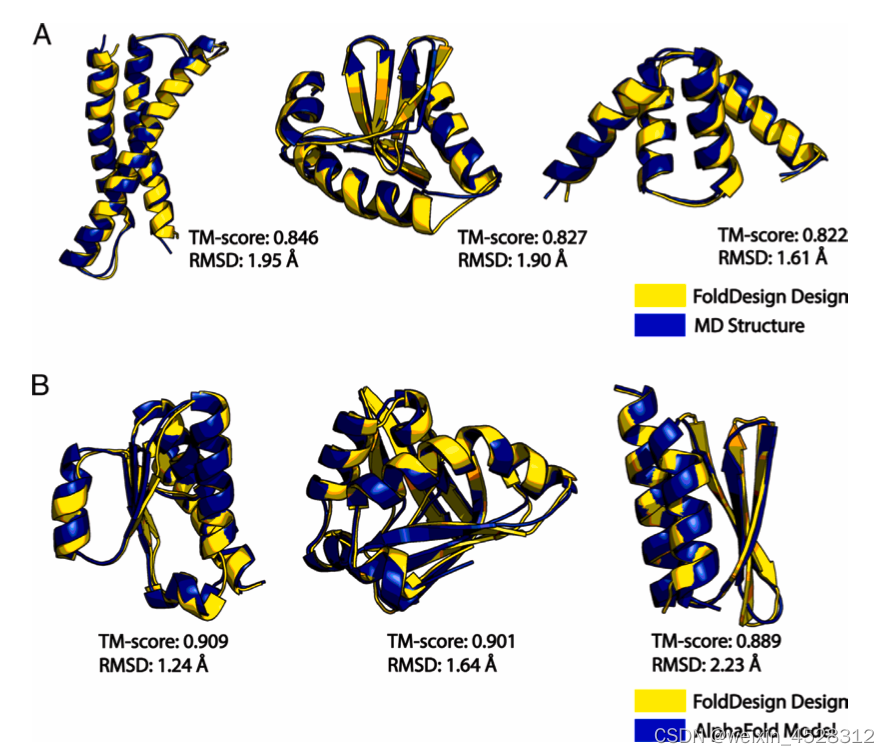
图6. FoldDesign设计的新结构以及与AlphaFold2结构预测比较示例
探索设计天然蛋白折叠之外的构型新空间
特别值得一提的是,尽管FoldDesign产生的折叠构型很好地保持了初始二级结构并且具有很高的稳定性(图5A-B),仍有约22%的FoldDesign构型与PDB数据库中的结构完全不同,即TM-score <0.5(图5E)。这些结果表明,FoldDesign不仅能够生成紧凑而稳定的结构,而且可以探索新的折叠空间。显然,这对于利用计算机来设计全新结构和功能的蛋白质至关重要。
为什么FoldDesign拥有设计全新拓扑结构的能力?为了回答这个问题,作者统计了自然界中所有已知5万1千多个非冗余蛋白质的超二级结构(也叫做‘Smotif’,即由一个loop链接两个连续二级结构的局域构型)。按照Smotif在自然蛋白中出现的频率,作者把这些超二级结构分成‘低频’(极少出现,频率低于0.001),‘次低频’(较少出现,低于0.01),‘次高频’(经常出现,低于0.1),和‘高频’(极常出现,高于0.1)等四个区域。
图7显示,FoldDesign在‘低频’和‘次低频’两个低频区的超二级结构采样远高于天然蛋白质。这一方面揭示出FoldDesign产生新拓扑构型的来源,即FoldDesign采用了与天然结构不同的超二级结构转角分布。另一方面来讲,因为FoldDesign的构型是计算机在给定物理力场中能量最优的结果,它本身没有类似于天然蛋白的进化压力。因此,这些数据也显示出,目前我们所看到的大自然蛋白质的有限构型(包括特殊的超二级结构的分布),应该主要是功能进化压力的结果。而FoldDesign这样没有进化压力的计算机蛋白质模拟,有助于探索自然界中没有产生的新的蛋白质结构和功能空间。
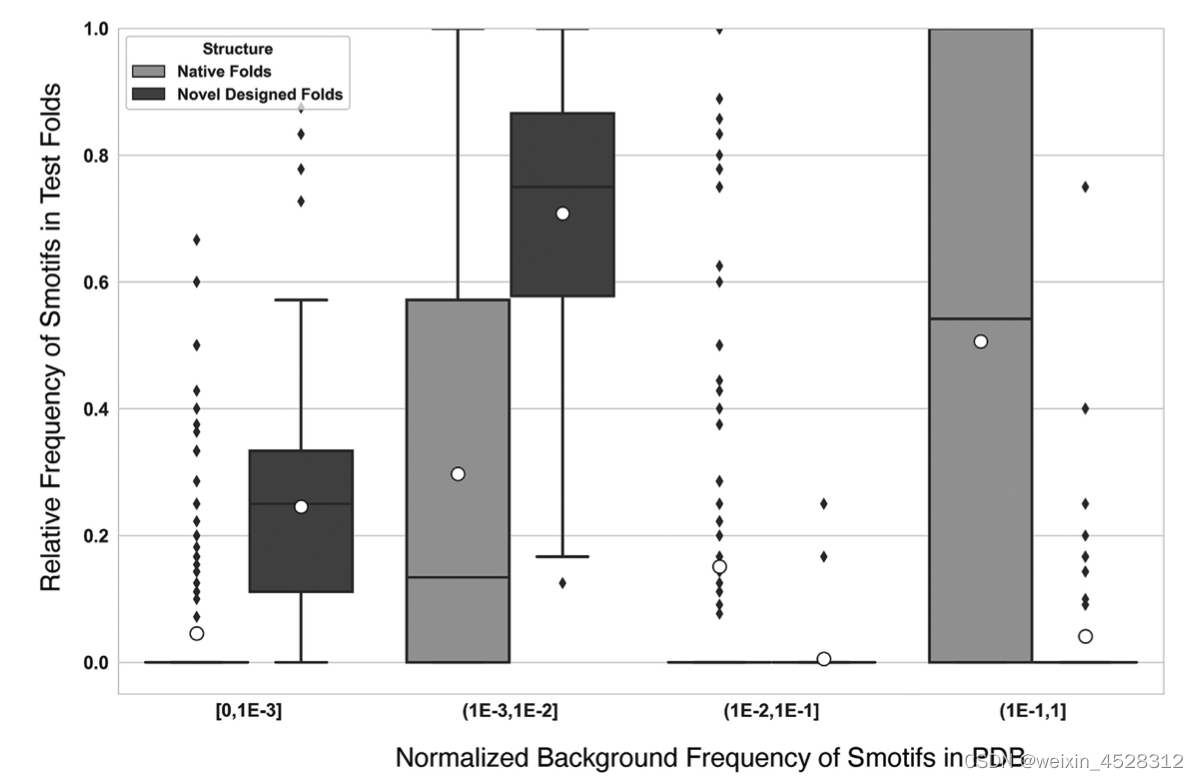
图7. FoldDesign结构与天然蛋白的超二级结构频率比较
图8显示了两个例子,其二级结构分布分别取自于PhoQ组氨酸激活酶和谷氨酸棒杆菌调节蛋白。虽然使用了与天然蛋白相同的二级结构类,但是因为FoldDesign采用了很多不常见的超二级结构,它最终设计的蛋白质整体结构与天然结构完全不同(最接近的TM-score只有0.467和0.451)。
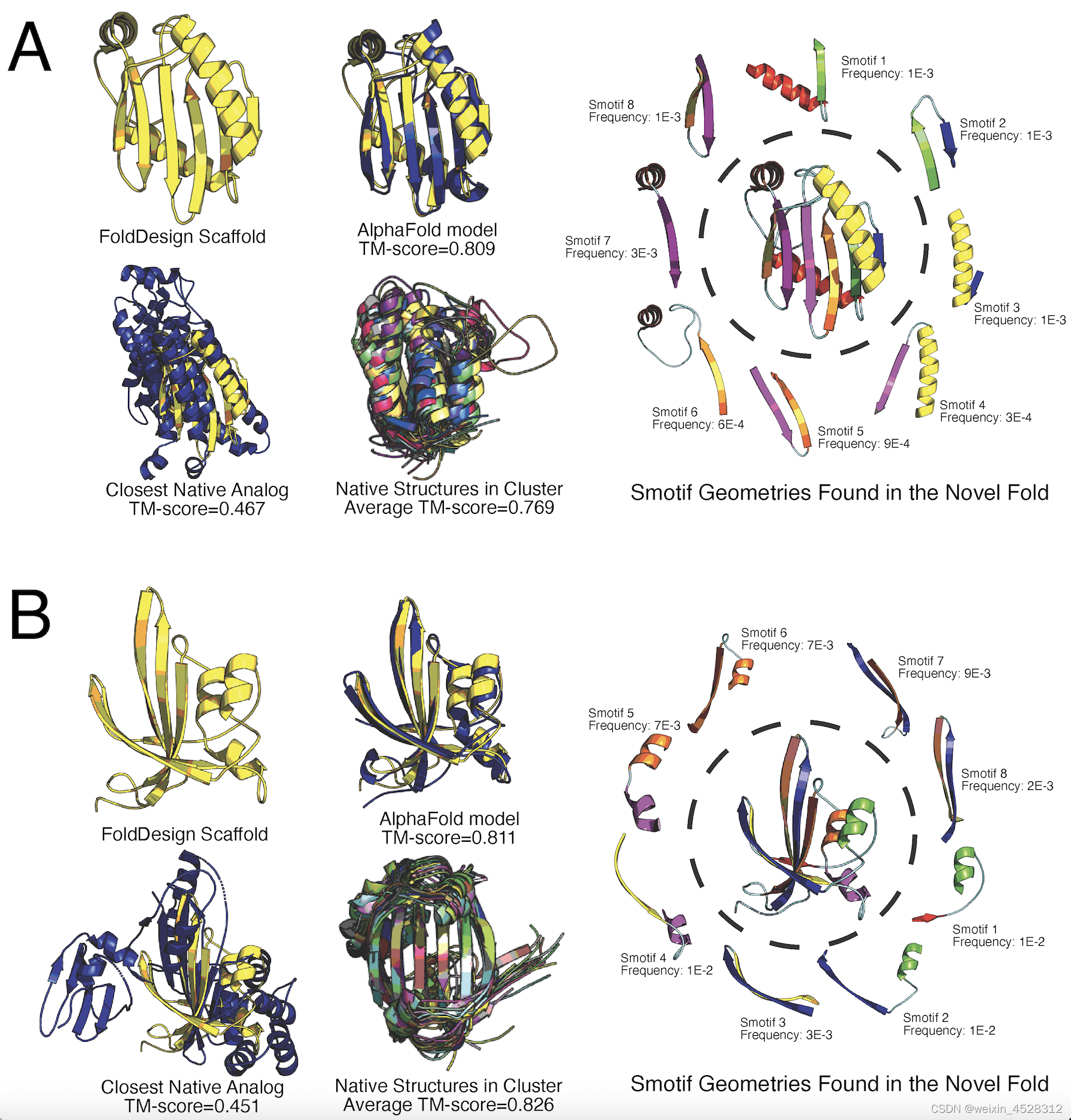
图8. 因为采用了不同的超二级结构(图右),FoldDesign设计出与PDB库天然蛋白完全不同的拓扑学构型(图左)。两个例子分别来自(A)PhoQ组氨酸激活酶(PDBID:1id0A)和(B)谷氨酸棒杆菌调节蛋白(PDBID:2p19A)
结论与展望
总之,本研究开发了一个新的蛋白结构从头设计算法(FoldDesign)。通过利用无序列片段组装模拟,FoldDesign能够对任何拓扑学构型,设计产生稳定紧致、高保度的蛋白质三级结构。大规模基准测试表明,与其他蛋白设计软件(如Rosetta)相比,FoldDesign设计能明显更好地复现人工所要求的蛋白质二级结构,其所设计的整体折叠构型也更紧凑、物理学上更加稳定。基于人工智能的结构预测实验显示,FoldDesign结构设计的蛋白序列能更大程度地折叠成FoldDesign产生的初始结构,表明FoldDesign构型具有更好的可设计性和自洽性。
特别重要的是,因为FoldDeign的构型完全基于物理势能优化,从而避免了自然进化的压力,其所设计的蛋白质倾向于采用自然界不常用的超二级结构,因此可能具有与自然蛋白完全不同的整体构型。这种新构型设计的能力对于探索蛋白世界的整体结构和功能空间,乃至设计产生新的生命体,都具有重要意义。
参考文献

