
热门标签
热门文章
- 1UniPro、PingCode、禅道,CTO如何选择合适的项目管理软件_禅道和pingcode对比
- 2代码随想录算法训练营第37天| 738.单调递增的数字、968.监控二叉树
- 3TIDB 初级课程体验 7 (用户管理与权限,跳过密码)
- 4Android Studio引入framework.jar包
- 5mysql 单行函数
- 6谷歌浏览器没有添加flash选项_电脑谷歌浏览器没有flash设置
- 7将xml文件转yolov5训练数据txt标签文件_xml转yolo
- 8IO流详解及常用方法
- 9sample gpt 无限长上下文
- 10Python如何pip批量安装指定包 - 最简单方法_pip install 多个包
当前位置: article > 正文
数据分析:甲基化分析-从DNA methylation的IDAT文件到CpG site的Beta values
作者:不正经 | 2024-04-28 01:00:25
赞
踩
数据分析:甲基化分析-从DNA methylation的IDAT文件到CpG site的Beta values
介绍
DNA Methylation和疾病的发生发展存在密切相关,它一般通过CH3替换碱基5‘碳的H原子,进而调控基因的转录。常用的DNA methylation是Illumina Infinium methylation arrays,该芯片有450K和850K(也即是EPIC)。
该脚本基于命令行模式运行:主要分为以下几部分
-
读取IDAT(甲基化芯片数据格式)数据和建立RGset对象;
-
质控(根据QC结果质控和根据强度P值小于0.01或0.05过滤);
-
标准化
-
过滤:
-
检测P值过滤;
-
去重复;
-
去除X和Y染色体的甲基化;
-
去除CpG岛的SNPs;
-
去除多比对的甲基化
- 输出matrix
准备文件
- Sample.csv: csv格式的table

- DNA methylation folder
IDAT/ ├── 202250800017 │ ├── 202250800017_R01C01_Grn.idat │ ├── 202250800017_R01C01_Red.idat │ ├── 202250800017_R02C01_Grn.idat │ ├── 202250800017_R02C01_Red.idat │ ├── 202250800017_R03C01_Grn.idat │ ├── 202250800017_R03C01_Red.idat │ ├── 202250800017_R04C01_Grn.idat │ ├── 202250800017_R04C01_Red.idat │ ├── 202250800017_R05C01_Grn.idat │ ├── 202250800017_R05C01_Red.idat │ ├── 202250800017_R06C01_Grn.idat │ ├── 202250800017_R06C01_Red.idat │ ├── 202250800017_R07C01_Grn.idat │ ├── 202250800017_R07C01_Red.idat │ ├── 202250800017_R08C01_Grn.idat │ └──202250800017_R08C01_Red.idat ├── 202250800184 │ ├── 202250800184_R01C01_Grn.idat │ ├── 202250800184_R01C01_Red.idat │ ├── 202250800184_R02C01_Grn.idat │ ├── 202250800184_R02C01_Red.idat │ ├── 202250800184_R03C01_Grn.idat │ ├── 202250800184_R03C01_Red.idat │ ├── 202250800184_R04C01_Grn.idat │ ├── 202250800184_R04C01_Red.idat │ ├── 202250800184_R05C01_Grn.idat │ ├── 202250800184_R05C01_Red.idat │ ├── 202250800184_R06C01_Grn.idat │ ├── 202250800184_R06C01_Red.idat │ ├── 202250800184_R07C01_Grn.idat │ ├── 202250800184_R07C01_Red.idat │ ├── 202250800184_R08C01_Grn.idat │ └── 202250800184_R08C01_Red.idat

- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- cross reactive probes files
850K: 13059_2016_1066_MOESM1_ESM_cross-reactive_probes.csv
450K: 48639-non-specific-probes-Illumina450k.csv
- 1
- 2
loading R packages
library(dplyr)
library(limma)
library(minfi)
library(missMethyl)
library(minfiData)
library(stringr)
library(ENmix)
library(RColorBrewer)
library(optparse)
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
get options
option_list = list(
make_option(c("-f", "--folder"), type="character", default=".",
help="folder with idat files [default= %default]", metavar="character"),
make_option(c("-t", "--target"), type="character", default=".",
help="target file with sample information [default= %default]", metavar="character"),
make_option(c("-a", "--array"), type="character", default=".",
help="the type of DNA chip array file [default= %default]", metavar="character"),
make_option(c("-o", "--out"), type="character", default="out",
help="output directory name [default= %default]", metavar="character")
);
opt_parser <- OptionParser(option_list=option_list);
opt <- parse_args(opt_parser);
print(opt)
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
Creating output directory
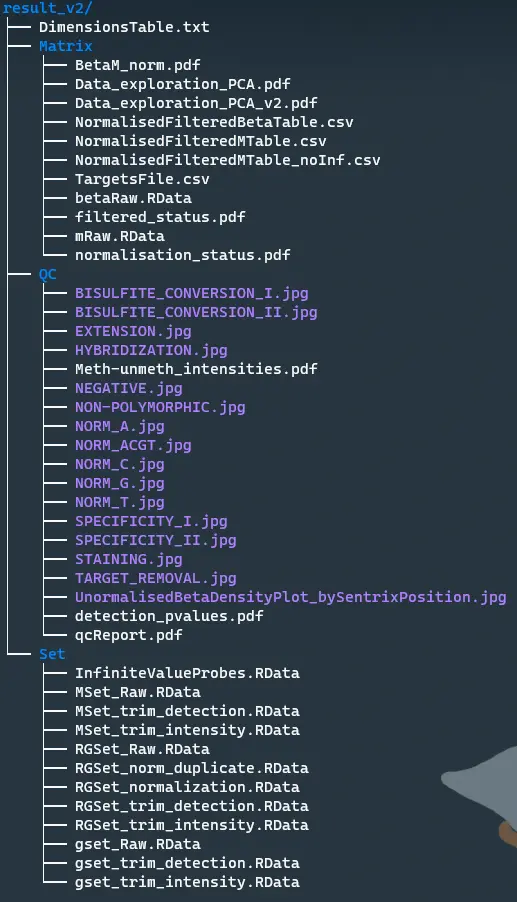
create_dir <- function(dirpath){ if(!dir.exists(dirpath)){ dir.create(dirpath, recursive = TRUE) } } # current dir current_dir <- getwd() # Result & QC dir out_dir <- opt$out qc_dir <- paste0(out_dir, "/QC") create_dir(qc_dir) # DataSet dir set_dir <- paste0(out_dir, "/Set") create_dir(set_dir) # Matrix dir raw_dir <- paste0(out_dir, "/Matrix") create_dir(raw_dir)
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
Chip Array annotation and cross reactive files
if(opt$chip == "850k"){
# For HumanMethylationEPIC bead chip array files (850k) load the following two packages and two files
library(IlluminaHumanMethylationEPICmanifest)
library(IlluminaHumanMethylationEPICanno.ilm10b4.hg19)
ann <- getAnnotation(IlluminaHumanMethylationEPICanno.ilm10b4.hg19)
crossreac_probes <- read.csv("/disk/user/zouhua/pipeline/methylseq_IDAT/util/13059_2016_1066_MOESM1_ESM_cross-reactive_probes.csv")
}else if(opt$chip == "450k"){
# For HumanMethylation450 bead chip array files (450k) load the following packages and two files
library(IlluminaHumanMethylation450kmanifest)
library(IlluminaHumanMethylation450kanno.ilmn12.hg19)
ann <- getAnnotation(IlluminaHumanMethylation450kanno.ilmn12.hg19)
crossreac_probes <- read.csv("/disk/user/zouhua/pipeline/methylseq_IDAT/util/48639-non-specific-probes-Illumina450k.csv")
}
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
Reading idat files
# list the files
list.files(opt$folder, recursive = TRUE)
# read in the sample sheet for the experiment
targets <- read.csv(opt$target)
head(targets)
- 1
- 2
- 3
- 4
- 5
Creating RGset files
RGSet <- read.metharray.exp(targets = targets)
targets$ID <- paste(targets$Sample_Name)
sampleNames(RGSet) <- targets$ID
print(RGSet)
- 1
- 2
- 3
- 4
Quality Checks
# C.1. Plot quality control plots (package ENmix) setwd(qc_dir) plotCtrl(RGSet) # C.2. Make PDF QC report (package minfi) # To include colouring samples by variable such as sentrix position include argument: sampGroups=targets$sentrix_pos setwd(current_dir) qcReport_pdf <- paste0(qc_dir, "/qcReport.pdf") qcReport(RGSet, sampNames = targets$Sample_Name, pdf = qcReport_pdf) # C.3. Make pre-normalisation beta density plots # Here samples are coloured by sentrix position nb.levels <- length(unique(targets$Array)) mycolors <- colorRampPalette(brewer.pal(8, "Dark2"))(nb.levels) jpeg(paste0(qc_dir, "/UnormalisedBetaDensityPlot_bySentrixPosition.jpg"), width = 800, height = 800) densityPlot(RGSet, sampGroups = targets$Array, pal = mycolors, ylim = c(0, 5)) dev.off() # C.4. Create MethylSet, RatioSet, then a GenomicRatioSet for further QC MSet <- preprocessRaw(RGSet) ratioSet <- ratioConvert(MSet, what = "both", keepCN = TRUE) gset <- mapToGenome(ratioSet, mergeManifest = T) # C.5. Perform QC on MSet and plot methylated versus unmethylated intensities qc <- getQC(MSet) pdf(paste0(qc_dir, "/Meth-unmeth_intensities.pdf"), height = 4, width = 4) par(mfrow = c(1, 1), family = "Times", las = 1) plotQC(qc) # If U and/or M intensity log medians are <10.5, sample is by default of bad quality dev.off() # C.6. remove any samples with meth or unmeth intensity < 10.5, then remove from all previous files if(sum(rowMeans(as.data.frame(qc)) < 10.5 ) > 0) { print(paste("Warning: sample", rownames(qc)[rowMeans(as.data.frame(qc)) < 10.5], "has mean meth-unmeth signal intensity <10.5. Remove sample.")) } keep.samples <- apply(as.data.frame(qc), 1, mean) > 10.5 RGSet_remain <- RGSet[, keep.samples] MSet_remain <- MSet[, keep.samples] ratioSet_remain <- ratioSet[, keep.samples] gset_remain <- gset[, keep.samples] targets_remain <- targets[keep.samples, ] ############################################################### ######## Calculate detection p values and plot ######## ############################################################### # Here the samples are coloured by sentrix ID to check for poor performing chips detP <- detectionP(RGSet_remain) pdf(paste0(qc_dir, "/detection_pvalues.pdf"), height = 4, width = 4) par(mfrow = c(1, 1), family = "Times", las = 1) barplot(colMeans(detP), col = as.numeric(factor(targets$Slide)), las = 2, cex.names = 0.8, ylim = c(0, 0.05), ylab = "Mean detection p-values") abline(h = 0.01 ,col = "red") dev.off() if(sum(colMeans(detP) > 0.01) > 0){ print(paste("Warning: sample", names(colMeans(detP))[colMeans(detP) > 0.01], "has >0.01 mean detection p-value. Remove sample")) } # If required: remove any samples with detection p value > 0.01, then remove from all previous files keep.samples_v2 <- apply(detP, 2, mean) < 0.01 RGSet_remain_v2 <- RGSet_remain[, keep.samples_v2] MSet_remain_v2 <- MSet_remain[, keep.samples_v2] ratioSet_remain_v2 <- ratioSet_remain[, keep.samples_v2] gset_remain_v2 <- gset_remain[, keep.samples_v2] targets_remain_v2 <- targets_remain[keep.samples_v2, ] glimpse(targets_remain_v2)
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- 44
- 45
- 46
- 47
- 48
- 49
- 50
- 51
- 52
- 53
- 54
- 55
- 56
- 57
- 58
- 59
- 60
- 61
- 62
- 63
- 64
- 65
- 66
- 67
- 68
- 69
- 70
output raw set files
save(RGSet, file = paste0(set_dir, "/RGSet_Raw.RData"))
save(MSet, file = paste0(set_dir, "/MSet_Raw.RData"))
save(gset, file = paste0(set_dir, "/gset_Raw.RData"))
save(RGSet_remain, file = paste0(set_dir, "/RGSet_trim_intensity.RData"))
save(MSet_remain, file = paste0(set_dir, "/MSet_trim_intensity.RData"))
save(gset_remain, file = paste0(set_dir, "/gset_trim_intensity.RData"))
save(RGSet_remain_v2, file = paste0(set_dir, "/RGSet_trim_detection.RData"))
save(MSet_remain_v2, file = paste0(set_dir, "/MSet_trim_detection.RData"))
save(gset_remain_v2, file = paste0(set_dir, "/gset_trim_detection.RData"))
size.RGSet_remain_v2 <- as.data.frame(t(dim(RGSet_remain_v2)))
size.MSet_remain_v2 <- as.data.frame(t(dim(MSet_remain_v2)))
size.gset_remain_v2 <- as.data.frame(t(dim(gset_remain_v2)))
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
Create raw unormalised and unfiltered beta table for later comparison
betaRaw <- getBeta(gset_remain_v2)
mRaw <- getM(gset_remain_v2)
save(betaRaw, file = paste0(raw_dir, "/betaRaw.RData"))
save(mRaw, file = paste0(raw_dir, "/mRaw.RData"))
- 1
- 2
- 3
- 4
- 5
Normalization
RGSet_norm <- preprocessFunnorm(RGSet_remain_v2)
size.RGSet_norm <- as.data.frame(t(dim(RGSet_norm)))
save(RGSet_norm, file = paste0(set_dir, "/RGSet_normalization.RData"))
# visualise what the data looks like before and after normalisation
pdf(paste0(raw_dir, "/normalisation_status.pdf"), height = 4, width = 4)
par(mfrow=c(1,2))
densityPlot(RGSet_remain_v2, sampGroups=targets$Sample_Source, main="Raw", legend=FALSE)
legend("top", legend = levels(factor(targets$Sample_Source)),
text.col=brewer.pal(8,"Dark2"))
densityPlot(getBeta(RGSet_norm), sampGroups=targets$Sample_Source,
main="Normalized", legend=FALSE)
legend("top", legend = levels(factor(targets$Sample_Source)),
text.col=brewer.pal(8, "Dark2"))
dev.off()
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
Data exploration
# MDS plots to look at largest sources of variation pal <- brewer.pal(8,"Dark2") pdf(paste0(raw_dir, "/Data_exploration_PCA.pdf"), height = 4, width = 4) par(mfrow=c(1, 2)) plotMDS(getM(RGSet_norm), top=1000, gene.selection="common", col=pal[factor(targets$Sample_Group)]) legend("top", legend=levels(factor(targets$Sample_Group)), text.col=pal, bg="white", cex=0.7) plotMDS(getM(RGSet_norm), top=1000, gene.selection="common", col=pal[factor(targets$Sample_Source)]) legend("top", legend=levels(factor(targets$Sample_Source)), text.col=pal, bg="white", cex=0.7) dev.off() # Examine higher dimensions to look at other sources of variation pdf(paste0(raw_dir, "/Data_exploration_PCA_v2.pdf"), height = 4, width = 6) par(mfrow=c(1, 3)) plotMDS(getM(RGSet_norm), top=1000, gene.selection="common", col=pal[factor(targets$Sample_Group)], dim=c(1,3)) legend("top", legend=levels(factor(targets$Sample_Group)), text.col=pal, cex=0.7, bg="white") plotMDS(getM(RGSet_norm), top=1000, gene.selection="common", col=pal[factor(targets$Sample_Group)], dim=c(2,3)) legend("topleft", legend=levels(factor(targets$Sample_Group)), text.col=pal, cex=0.7, bg="white") plotMDS(getM(RGSet_norm), top=1000, gene.selection="common", col=pal[factor(targets$Sample_Group)], dim=c(3,4)) legend("topright", legend=levels(factor(targets$Sample_Group)), text.col=pal, cex=0.7, bg="white") dev.off()
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
filtering
# ensure probes are in the same order in the mSetSq and detP objects detP <- detP[match(featureNames(RGSet_norm), rownames(detP)), ] # remove any probes that have failed in one or more samples keep <- rowSums(detP < 0.01) == ncol(RGSet_norm) table(keep) RGSet_normFlt <- RGSet_norm[keep, ] RGSet_normFlt # removing duplicated pData <- pData(RGSet_normFlt) pData2 <- pData[order(pData(RGSet_normFlt)$Sample_Name), ] dim(pData2) head(pData2) dups <- pData2$Sample_Name[duplicated(pData2$Sample_Name)] if( length(dups) > 0 ){ whdups <- lapply(dups, function(x){which(pData2$Sample_Name == x)}) whdups2rem <- sapply(1:length(dups), function(i) rbinom(1, 1, prob = 1/length(whdups[[1]])))+1 torem <- sapply(1:length(whdups), function(i){whdups[[i]][whdups2rem[i]]}) pData2 <- pData2[-torem, ] } RGSet_norm_duplicate <- RGSet_normFlt[, rownames(pData2)] save(RGSet_norm_duplicate, file = paste0(set_dir, "/RGSet_norm_duplicate.RData")) size.detP <- as.data.frame(t(dim(RGSet_norm_duplicate))) # if your data includes males and females, remove probes on the sex chromosomes keep <- !(featureNames(RGSet_norm_duplicate) %in% ann$Name[ann$chr %in% c("chrX","chrY")]) table(keep) RGSet_norm_duplicate <- RGSet_norm_duplicate[keep, ] # remove probes with SNPs at CpG site RGSetFlt <- dropLociWithSnps(RGSet_norm_duplicate) RGSetFlt # exclude cross reactive probes keep <- !(featureNames(RGSetFlt) %in% crossreac_probes$TargetID) table(keep) RGSetFlt <- RGSetFlt[keep,] RGSetFlt pdf(paste0(raw_dir, "/filtered_status.pdf"), height = 4, width = 4) par(mfrow=c(1, 2)) plotMDS(getM(RGSet_norm), top=1000, gene.selection="common", main="Normalized", col=pal[factor(targets$Sample_Source)]) legend("top", legend=levels(factor(targets$Sample_Source)), text.col=pal, bg="white", cex=0.7) plotMDS(getM(RGSetFlt), top=1000, gene.selection="common", main="Normalized_filter", col=pal[factor(targets$Sample_Source)]) legend("top", legend=levels(factor(targets$Sample_Source)), text.col=pal, bg="white", cex=0.7) dev.off()
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- 44
- 45
- 46
- 47
- 48
- 49
- 50
- 51
- 52
- 53
Normalization matrix
betaNorm <- getBeta(RGSetFlt) dim(betaNorm) size.betaNorm <- as.data.frame(t(dim(betaNorm))) mNorm <- getM(RGSetFlt) dim(mNorm) size.mNorm <- as.data.frame(t(dim(mNorm))) # visualization pdf(paste0(raw_dir, "/filtered_status.pdf"), height = 4, width = 4) par(mfrow=c(1,2)) densityPlot(betaNorm, sampGroups=targets$Sample_Group, main="Beta values", legend=FALSE, xlab="Beta values") legend("top", legend = levels(factor(targets$Sample_Group)), text.col=brewer.pal(8,"Dark2")) densityPlot(mNorm, sampGroups=targets$Sample_Group, main="M-values", legend=FALSE, xlab="M values") legend("topleft", legend = levels(factor(targets$Sample_Group)), text.col=brewer.pal(8,"Dark2")) dev.off() # Check for NA and -Inf values betaRaw.na <- betaRaw[!complete.cases(betaRaw), ] dim(betaRaw.na) NAbetas <- rownames(betaRaw.na) betaNAsNorm <- betaNorm[rownames(betaNorm) %in% NAbetas, ] # Should be [1] 0 x ncol(betaNorm) # Check for infinite values in m table, replace in the no infinite values m table TestInf <- which(apply(mNorm, 1, function(i)sum(is.infinite(i))) > 0) save(TestInf, file = paste0(set_dir, "/InfiniteValueProbes.RData")) mNoInf <- mNorm mNoInf[!is.finite(mNoInf)] <- min(mNoInf[is.finite(mNoInf)]) TestInf2 <- which(apply(mNoInf, 1, function(i) sum(is.infinite(i))) > 0) TestInf2 # Should be named integer(0) size.mNoInf <- as.data.frame(t(dim(mNoInf))) # Write tables write.table(targets_remain_v2, file=paste0(raw_dir, "/TargetsFile.csv"), sep=",", col.names=NA) write.table(betaNorm, file=paste0(raw_dir, "/NormalisedFilteredBetaTable.csv"), sep=",", col.names=NA) write.table(mNorm, file=paste0(raw_dir, "/NormalisedFilteredMTable.csv"), sep=",", col.names=NA) write.table(mNoInf, file=paste0(raw_dir, "/NormalisedFilteredMTable_noInf.csv"), sep=",", col.names=NA)
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
Create dimensions table
DimensionsTable <- rbind(size.RGSet_remain_v2, size.MSet_remain_v2, size.gset_remain_v2,
size.RGSet_norm, size.detP,
size.betaNorm, size.mNorm, size.mNoInf)
rownames(DimensionsTable) <- c("RGset_size", "MSet_size", "gset_size",
"fun_size", "detP_lost",
"beta_size", "m_size", "mNoInf_size")
colnames(DimensionsTable) <- c("Probe_number", "Sample_number")
write.table(DimensionsTable, file=paste0(out_dir, "/DimensionsTable.txt"), sep="\t", col.names=NA)
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
运行
Rscript DNA_Methylation.R -f IDAT -t Sample.csv -a 850k -o result_v2
- 1
Software versions
sessionInfo()
- 1
R version 4.0.2 (2020-06-22) Platform: x86_64-conda_cos6-linux-gnu (64-bit) Running under: CentOS Linux 8 (Core) Matrix products: default BLAS/LAPACK: /disk/share/anaconda3/lib/libopenblasp-r0.3.10.so locale: [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=en_US.UTF-8 [4] LC_COLLATE=en_US.UTF-8 LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8 [7] LC_PAPER=en_US.UTF-8 LC_NAME=C LC_ADDRESS=C [10] LC_TELEPHONE=C LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C attached base packages: [1] stats4 parallel stats graphics grDevices utils datasets methods base other attached packages: [1] RColorBrewer_1.1-2 ENmix_1.26.9 [3] doParallel_1.0.16 stringr_1.4.0 [5] minfiData_0.36.0 IlluminaHumanMethylation450kmanifest_0.4.0 [7] missMethyl_1.24.0 IlluminaHumanMethylationEPICanno.ilm10b4.hg19_0.6.0 [9] IlluminaHumanMethylation450kanno.ilmn12.hg19_0.6.0 minfi_1.36.0 [11] bumphunter_1.32.0 locfit_1.5-9.4 [13] iterators_1.0.13 foreach_1.5.1 [15] Biostrings_2.58.0 XVector_0.30.0 [17] SummarizedExperiment_1.20.0 Biobase_2.50.0 [19] MatrixGenerics_1.2.1 matrixStats_0.58.0 [21] GenomicRanges_1.42.0 GenomeInfoDb_1.26.4 [23] IRanges_2.24.1 S4Vectors_0.28.1 [25] BiocGenerics_0.36.0 limma_3.46.0 [27] dplyr_1.0.5 loaded via a namespace (and not attached): [1] AnnotationHub_2.22.0 BiocFileCache_1.14.0 plyr_1.8.6 [4] splines_4.0.2 BiocParallel_1.24.1 digest_0.6.27 [7] htmltools_0.5.1.1 RPMM_1.25 fansi_0.4.2 [10] magrittr_2.0.1 memoise_2.0.0 cluster_2.1.0 [13] readr_1.4.0 annotate_1.68.0 askpass_1.1 [16] siggenes_1.64.0 lpSolve_5.6.15 prettyunits_1.1.1 [19] blob_1.2.1 rappdirs_0.3.3 xfun_0.20 [22] crayon_1.4.1 RCurl_1.98-1.3 jsonlite_1.7.2 [25] genefilter_1.72.0 GEOquery_2.58.0 impute_1.64.0 [28] survival_3.2-10 glue_1.4.2 zlibbioc_1.36.0 [31] DelayedArray_0.16.3 irr_0.84.1 Rhdf5lib_1.12.1 [34] HDF5Array_1.18.1 DBI_1.1.1 rngtools_1.5 [37] Rcpp_1.0.6 xtable_1.8-4 progress_1.2.2 [40] bit_4.0.4 mclust_5.4.7 preprocessCore_1.52.1 [43] httr_1.4.2 gplots_3.1.1 ellipsis_0.3.1 [46] pkgconfig_2.0.3 reshape_0.8.8 XML_3.99-0.6 [49] sass_0.3.1 dbplyr_2.1.1 utf8_1.2.1 [52] dynamicTreeCut_1.63-1 later_1.1.0.1 tidyselect_1.1.0 [55] rlang_0.4.10 AnnotationDbi_1.52.0 BiocVersion_3.12.0 [58] tools_4.0.2 cachem_1.0.4 generics_0.1.0 [61] RSQLite_2.2.5 ExperimentHub_1.16.0 evaluate_0.14 [64] fastmap_1.1.0 yaml_2.2.1 org.Hs.eg.db_3.12.0 [67] knitr_1.31 bit64_4.0.5 beanplot_1.2 [70] caTools_1.18.2 scrime_1.3.5 purrr_0.3.4 [73] nlme_3.1-150 doRNG_1.8.2 sparseMatrixStats_1.2.1 [76] mime_0.10 nor1mix_1.3-0 xml2_1.3.2 [79] biomaRt_2.46.3 compiler_4.0.2 rstudioapi_0.13 [82] interactiveDisplayBase_1.28.0 curl_4.3 tibble_3.1.0 [85] statmod_1.4.35 geneplotter_1.66.0 bslib_0.2.4 [88] stringi_1.4.6 GenomicFeatures_1.42.2 lattice_0.20-41 [91] Matrix_1.3-2 multtest_2.46.0 vctrs_0.3.7 [94] pillar_1.6.0 lifecycle_1.0.0 rhdf5filters_1.2.0 [97] BiocManager_1.30.12 jquerylib_0.1.3 data.table_1.14.0 [100] bitops_1.0-6 httpuv_1.5.5 rtracklayer_1.50.0 [103] R6_2.5.0 promises_1.2.0.1 KernSmooth_2.23-18 [106] codetools_0.2-18 gtools_3.8.2 MASS_7.3-53.1 [109] assertthat_0.2.1 rhdf5_2.34.0 openssl_1.4.3 [112] GenomicAlignments_1.26.0 Rsamtools_2.6.0 GenomeInfoDbData_1.2.4 [115] hms_1.0.0 quadprog_1.5-8 grid_4.0.2 [118] tidyr_1.1.3 base64_2.0 rmarkdown_2.7 [121] DelayedMatrixStats_1.12.3 illuminaio_0.32.0 shiny_1.6.0 [124] tinytex_0.31
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- 44
- 45
- 46
- 47
- 48
- 49
- 50
- 51
- 52
- 53
- 54
- 55
- 56
- 57
- 58
- 59
- 60
- 61
- 62
- 63
- 64
- 65
- 66
- 67
- 68
- 69
- 70
- 71
- 72
- 73
- 74
- 75
参考
声明:本文内容由网友自发贡献,不代表【wpsshop博客】立场,版权归原作者所有,本站不承担相应法律责任。如您发现有侵权的内容,请联系我们。转载请注明出处:https://www.wpsshop.cn/w/不正经/article/detail/499634
推荐阅读
相关标签

