
- 1通达信 day文件 解析_Flink内置数据源:文件数据源
- 2腾讯云4核8G服务器支持多少人在线访问?
- 3springBoot 启动指定配置文件环境多种方案_springboot指定配置文件启动
- 4【远程桌面】nomachine下载安装使用教程、zerotier下载安装使用教程超详细
- 5阿里云抢占式实例服务器优缺点适用场景及收费标准_抢占式服务器是什么
- 6【MATLAB】tvf_emd_ MFE_SVM_LSTM 神经网络时序预测算法
- 7基于Python淘宝图书销售数据可视化系统设计与实现(Django框架) 研究背景与意义、国内外研究现状
- 8【知识图谱】Py2neo操作Neo4j使用教程_py2neo neo4j 5.12
- 9吴恩达AI FOR Everyone|人工智能入门笔记|_ai for everyone 1-35
- 102024年阿里云2核4G配置服务器测评_ECS和轻量性能测评
玩转ChatGPT:R代码Debug一例_chatgpt debug
赞
踩
一、写在前面
今天家里领导发来求助,说是用GPT-3.5写一个 计算mRNA干性指数 的R代码,运行报错。让我用GPT-4帮忙Debug一哈。
搞了半小时,还是有亿点感悟,写段文字记录记录。
二、踩坑过程
(1)先看原始的咒语
咒语:我已经使用synapser包计算得到了每个基因的干性权重标准值(第一列是基因名,第二列是权重值),现在需要计算TCGA中HCC的RNA-seq测序数据(第一行是样本名,第一列是基因名)每个样本的mRNA干性指数。
具体方法如下:
对于每个TCGA HCC测序数据中的样本,提取与基因mRNA干性权重标准值相同的基因的表达谱。通过Spearman相关系数计算每个样本的基因表达谱和基因mRNA干性权重标准值之间的相关性。对于每个样本,将其计算出的干性指数标准化到 [0,1] 的范围内。最后将所有样本的干性指数输出为一个表格。
输出的代码为:
- # 基因mRNA干性权重标准值文件的路径
-
- weight_file <- "干性权重.txt"
-
- # TCGA HCC测序数据文件的路径
-
- data_file <- "HTSeq - FPKM-mRNA.merge.txt"
-
- # 导入基因mRNA干性权重标准值
-
- weight <- read.table(weight_file, header = TRUE)
-
- # 导入TCGA HCC测序数据
-
- data <- read.csv(data_file, row.names = 1)
-
- # 提取与基因mRNA干性权重标准值相同的基因的表达谱
-
- common_genes <- intersect(rownames(weight), colnames(data))
-
- X <- data[, common_genes]
-
- # 计算每个样本的mRNA干性指数
-
- scores <- apply(X, 1, function(z) {
-
- cor(z, weight$Weight, method = "spearman", use = "complete.obs")
-
- })
-
- # 标准化干性指数到 [0,1] 范围内
-
- normalized_scores <- (scores - min(scores)) / (max(scores) - min(scores))
-
- # 构建结果表格
-
- result <- data.frame(sample_name = rownames(X), mRNAsi_score = normalized_scores)
-
- # 打印结果
-
- print(result)}
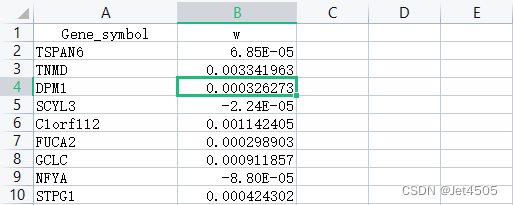
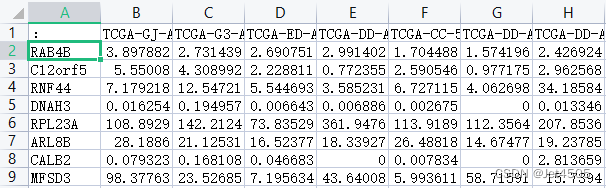
两个文件的数据格式如下(分别为干性权重.txt以及HTSeq - FPKM-mRNA.merge.txt):
RStudio运行以后报错如下:
Error in cor(z, weight$Weight, method = "spearman", use = "complete.obs") :要么同时设定'x'和'y',要么只设定矩阵式的'x'
由于我并不会R语言,我甚至不懂她这个算法是干啥的,所以就直接无脑丢给了GPT-4查查原因。
(2)先用GPT-4找错误
写一个咒语:运行代码:{XXX};
出现报错:{Error in cor(z, weight$Weight, method = "spearman", use = "complete.obs") : 要么同时设定'x'和'y',要么只设定矩阵式的'x'};
请问代码有什么错误?
输出如下:

主要变的是这:
- # 提取与基因mRNA干性权重标准值相同的基因的表达谱
-
- common_genes <- intersect(rownames(weight), colnames(data))
-
- X <- data[, common_genes]
-
- # 根据共享基因筛选权重
-
- selected_weight <- weight[rownames(weight) %in% common_genes, ]
-
- # 计算每个样本的mRNA干性指数
-
- scores <- apply(X, 1, function(z) {
-
- cor(z, weight$Weight, method = "spearman", use = "complete.obs")
-
- })
但是,依旧报错,也是同样的错误。
接着又告诉TA错误,GPT继续改进:

修改如下:
- # 根据共享基因筛选权重,并按照X的行名进行重排序
-
- selected_weight <- weight[rownames(weight) %in% common_genes, ]
-
- selected_weight <- selected_weight[match(rownames(X), rownames(selected_weight)), ]
还是没有用。
我开始怀疑是否是数据格式的问题,可能需要告诉GPT数据格式。
与此同时,家里领导也分享了想法,然后提供了一个修改方案:
- # 提取与基因mRNA干性权重标准值相同的基因的表达谱
-
- common_genes <- intersect(rownames(weight), rownames(data))
-
- X <- data[common_genes, ]
-
- w <- weight[common_genes]
但是还是报错。可以看到,其实都觉得是这一步出了问题,所以我打算进一步问GPT。
(3)优化咒语
咒语:运行下面代码:{XXX};
数据的格式为分别为:
weight_file为{Gene_symbol w
TSPAN6 6.85388903246113e-05},
data_file 为{Tags TCGA-GJ-A3OU-01 TCGA-G3-A7M9-01 TCGA-ED-A627-01 TCGA-DD-AACB-01 TCGA-CC-5264-01
RAB4B 3.897881937 2.731438761 2.690751371 2.991402482 1.704487843
};
报错:{XXX},何修改代码?

改的还是那一段:
- # 提取与基因mRNA干性权重标准值相同的基因的表达谱
-
- common_genes <- intersect(rownames(weight), rownames(data))
-
- X <- data[common_genes, ]
-
- # 根据共享基因筛选权重,并按照X的行名进行重排序
-
- w <- weight[rownames(weight) %in% common_genes, ]
-
- w <- w[match(rownames(X), rownames(w)), ]
依旧报错,我继续追问3-4回合,GPT也一直改代码,但是依旧行不通。
做到现在,可以排除数据格式的原因,代码应该也不是很难,但是为何报错呢?
没办法只能一步一步来解析了。
(4)解释整个代码
写一个咒语:请详细解析这个代码的功能,代码:{XXX}。

集中报错都在第二、三步骤,但我觉得GPT不至于连着简单的算法都不会,而且数据格式TA是知道的。
所以我考虑,会不会数据读取就有错误??!!
看了一下Rstudio的数据监视窗口,果然:
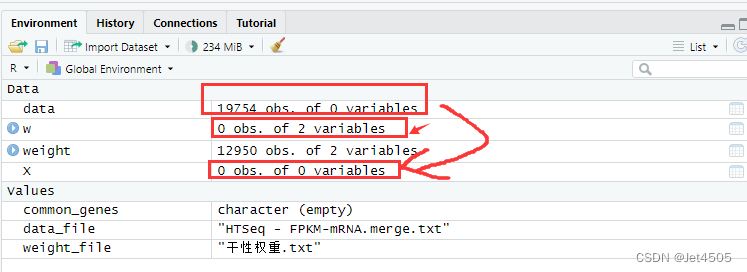
Data都没有数值,当然算不下去!!!
(5)优化数据读取代码
咒语:data_file 为{Tags TCGA-GJ-A3OU-01 TCGA-G3-A7M9-01 TCGA-ED-A627-01 TCGA-DD-AACB-01 TCGA-CC-5264-01
RAB4B 3.897881937 2.731438761 2.690751371 2.991402482 1.704487843
};读取代码为:data <- read.csv(data_file, row.names = 1),但是读取为0列,我需要全部列的信息,如何修改代码?

运行,数据读出来了:
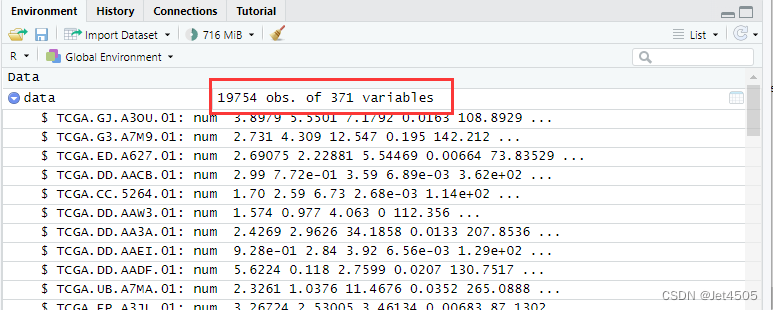
但是运行代码,依旧报错。
我有强烈预感,还是数据格式的问题,于是打开数据框,一目了然:
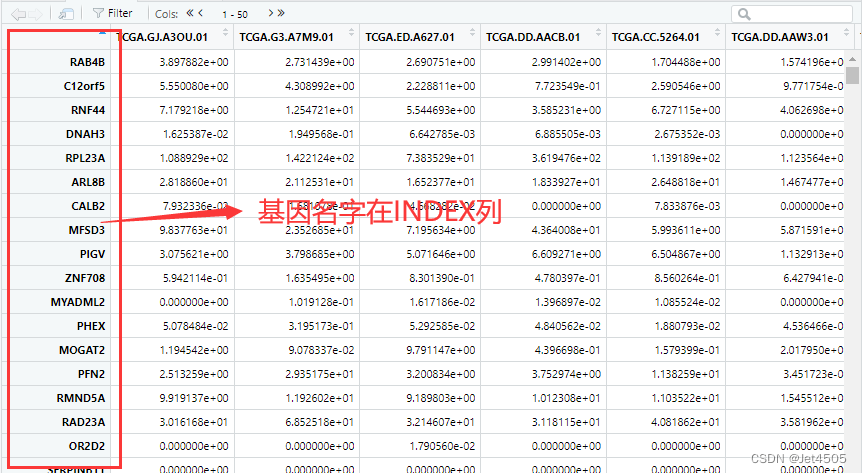
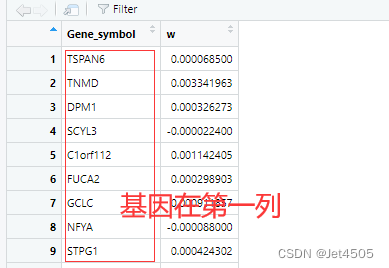
基因名所在的列都不一致,匹配不了很正常,这个代码我会改:
weight <- read.table(weight_file, header = TRUE, row.names = 1)这样,数据就对齐了:
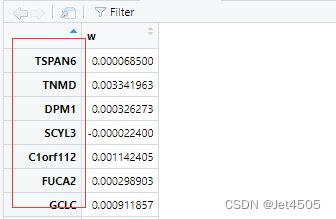
最后再运行代码(当然中间有些代码已经有修改,根据之前追问GPT的那几次),没报错了,至于结果对不对,我也不懂,至少跑通了。
万万没想到,居然是最不起眼的数据读取出问题,看来GPT-4也还是识别不出,一直在修改下面的算法核心部分。
下一步我测试下直接用GPT-4从头生成代码会如何?
(6)GPT-4生成代码
咒语:我已经使用synapser包计算得到了每个基因的干性权重标准值,现在需要计算TCGA中HCC的RNA-seq测序数据每个样本的mRNA干性指数。
具体方法如下:
对于每个TCGA HCC测序数据中的样本,提取与基因mRNA干性权重标准值相同的基因的表达谱。通过Spearman相关系数计算每个样本的基因表达谱和基因mRNA干性权重标准值之间的相关性。对于每个样本,将其计算出的干性指数标准化到 [0,1] 的范围内。最后将所有样本的干性指数输出为一个表格。
其中,干性权重标准值的数据格式为{Gene_symbol w
TSPAN6 6.85388903246113e-05},
TCGA中HCC的RNA-seq测序数据的格式为{Tags TCGA-GJ-A3OU-01 TCGA-G3-A7M9-01 TCGA-ED-A627-01 TCGA-DD-AACB-01 TCGA-CC-5264-01
RAB4B 3.897881937 2.731438761 2.690751371 2.991402482 1.704487843
};请用R语言写出代码。
代码如下:
- # 首先,安装和加载必要的包
-
- if (!require("dplyr")) {
-
- install.packages("dplyr")
-
- }
-
- library(dplyr)
-
- # 读取你的基因干性权重标准值数据
-
- dryness_weights <- read.table("dryness_weights.txt", header = TRUE, sep = "\t")
-
- # 读取你的TCGA HCC RNA-seq数据
-
- rna_seq_data <- read.table("rna_seq_data.txt", header = TRUE, sep = "\t")
-
- # 确保基因在两个数据框中都存在
-
- common_genes <- intersect(dryness_weights$Gene_symbol, rna_seq_data$Tags)
-
- # 提取共同基因的数据
-
- dryness_weights_filtered <- dryness_weights[dryness_weights$Gene_symbol %in% common_genes, ]
-
- rna_seq_data_filtered <- rna_seq_data[rna_seq_data$Tags %in% common_genes, ]
-
- # 计算每个样本的mRNA干性指数
-
- dryness_index <- apply(rna_seq_data_filtered[, -1], 2, function(sample_expr) {
-
- cor(dryness_weights_filtered$w, sample_expr, method = "spearman")
-
- })
-
- # 标准化干性指数到 [0,1] 的范围内
-
- dryness_index_normalized <- (dryness_index - min(dryness_index)) / (max(dryness_index) - min(dryness_index))
-
- # 输出结果
-
- dryness_index_df <- data.frame(sample_id = names(dryness_index_normalized), dryness_index = dryness_index_normalized)
-
- write.table(dryness_index_df, "dryness_index.txt", sep = "\t", quote = FALSE, row.names = FALSE)
跑出来的结果跟GPT-3.5的比,数量上都是一致的,共有371个数值,具体数值也一致:
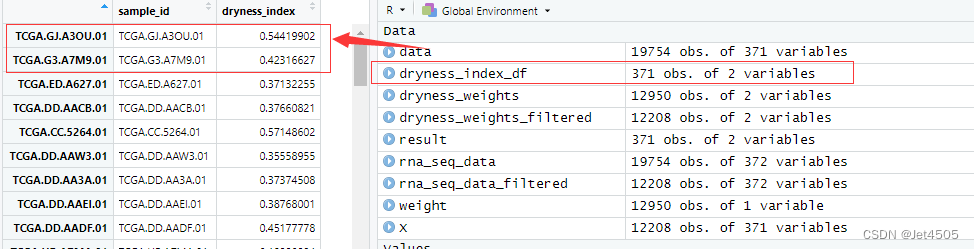
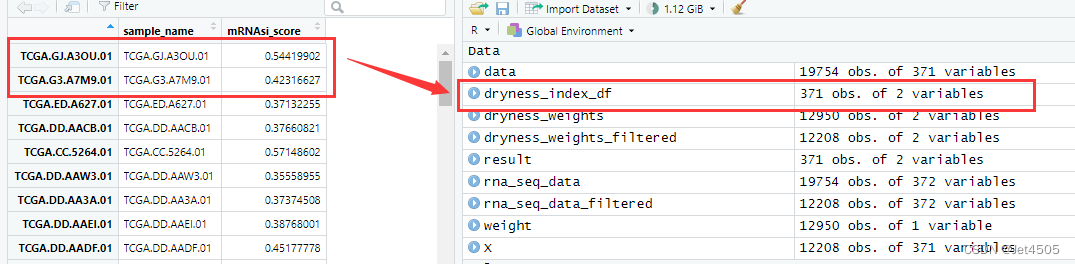
结束!同样的咒语我去试了GPT-3.5确实一次性写不出堪比GPT-4的代码。
三、写在最后
三点启示吧:
(1)在写咒语的时候,涉及到数据的,一定要直接复制你的数据格式给GPT(就直接复制粘贴,GPT能识别表格的分隔符),最好不要直接用文字描述,容易说不清楚;
(2)通过这个事例发现,GPT-4在找bug的时候,还是有些弱点。可能跟TA不能运行代码有关吧。然而,写代码能力还是强于GPT-3.5!
(3)最后,还是得自己理解代码,才能最高效的用GPT辅助Debug,偷懒不得啊!

