
- 1Apollo环境搭建_apollo9.0环境搭建
- 2芯片SIAT-002测试PCB板设计
- 3uwb定位算法c++代码_沃旭隧道/管廊人员定位技术方案
- 4antd vue Tabs控件的使用
- 5pandas实现按行取值或者按照行取另外一组数据的值_pandas读取excel数据并以其中一列作为字段的键值,并用其他列作为值
- 6Sqli-labs靶场第16关详解[Sqli-labs-less-16]自动化注入-SQLmap工具注入
- 7HarmonyOS应用开发者基础认证 试题及答案_下面哪一个事件方法可以获取到list滑动的偏移量
- 8UFUN API 函数_uf_ui_select_with_class_dialog
- 9Outlook2019一直提示输入用户名密码的解决方案_outlook2019一直弹出输入密码
- 10【历史上的今天】8 月 27 日:第一个面向对象编程语言创造者诞生;IE 衰亡起点;IBM研制世界最小计算机逻辑电路_第一个面向对象的语言
新国大张阳教授团队开发精度远超AlphaFold的AI蛋白质互作结构预测算法_深药科技张阳背景
赞
踩
蛋白质是一类由20种不同氨基酸组成的大分子有机化合物,它们是生命体内各种生物活动的最主要的执行者。蛋白质的绝大多数生物学功能都是通过与其它蛋白质的相互作用来实现。因此,蛋白质-蛋白质相互作用(简称‘蛋白质互作’)在生命功能的实现以及生物的进化过程中都扮演极其重要的角色。例如,抗体和抗原蛋白的相互作用,帮助生命个体识别和抵御外界病原体的入侵;受体和配体蛋白的相互作用,触发细胞信号传导通路;转录因子和共转录因子蛋白之间的相互作用,调节基因的转录和基因的修复;酶蛋白和底物的相互作用,催化新陈代谢的进程,等等。正是它们在细胞生物学上的这种特殊重要性,使得蛋白质互作成为许多现代药物设计的关键靶点。
尽管它们的重要性,如何确定蛋白质复合物的三维结构以及如何利用蛋白质互作的结构信息去指导蛋白质功能注解和药物设计,一直是现代生物医学的一个重要问题。在传统的结构生物学研究中,人们可以使用X射线晶体衍射、核磁共振及冷冻电镜等实验手段去解析蛋白质复合物的原子结构。但是结构生物学实验往往耗费大量人力物力,无法用于大规模获取蛋白质组级别的分子结构信息。并且,因为技术上的限制(如结构的稳定性以及高阶复合物的尺度等),某些蛋白质复合物的结构并不能通过传统的结构生物学实验手段获得。近年来,随着AlphaFold2等AI算法在蛋白质结构预测问题上的成功,通过计算手段获得高质量的蛋白质及复合物结构已经成为可能。但是,目前绝大多数AI算法都是通过对已知蛋白结构和基因进化关系的训练和学习来预测蛋白结构。而基因进化的信息主要从现有基因序列的多序列比对(MSA)来获得。因此,这些AI算法的实际预测精度,往往受目标蛋白MSA的质量影响较大,尤其在同源序列较少的孤儿蛋白(orphan proteins)和蛋白质复合物上的表现并不尽如人意。
近日,新加坡国立大学和美国密歇根大学张阳教授团队在顶级科学杂志《Nature Methods》上发表了题为“Improving deep learning protein monomer and complex structure prediction using DeepMSA2 with huge metagenomics data” 的论文。该论文研发了两款新的软件来提高蛋白质互作的结构预测精度。首先,作者开发了DeepMSA2,利用递推动态规划和隐马尔科夫模型算法,从海量宏基因组序列库中快速提取高质量MSA数据。然后利用新开发的DMFold软件构造蛋白质复合物的三维结构。实验结果显示,DMFold/DeepMSA2对蛋白质复合物的结构预测精度要显著优于AlphaFold2等算法。密歇根大学的郑伟博士为该论文的第一作者,张阳教授和P. Lydia Freddolino教授为论文共同通讯作者。

I. 背景介绍
在基于AI的蛋白质及其复合物结构预测中,一个重要的问题就是如何构建多重序列比对或者MSA。因为MSA中包含的基因和蛋白质的共进化信息,可以作为深度学习模型空间约束预测的特征输入。具体来讲,如果在进化过程中,蛋白质的其中一个氨基酸位点发生了突变,且破坏了氨基酸残基间的相互作用,则该蛋白会变得不稳定,拥有该突变的物种不容易存活下来。但是若与其有空间相互作用的氨基酸同时也发生了突变,且这两个突变又能够很好的作用在一起,使得蛋白质结构继续稳定下来,这种生物也就可以继续存活下来。这种现象被称为基因和蛋白质共进化(图1)。因为现存生命个体中的蛋白质都经过了生命长河共进化的洗礼,所以将现有海量蛋白序列进行多序列比对,可以有效推导出蛋白质共进化以及氨基酸空间距离的信息。因此,MSA及共进化信息被广泛应用于基于AI和深度学习的蛋白质三级结构预测中。
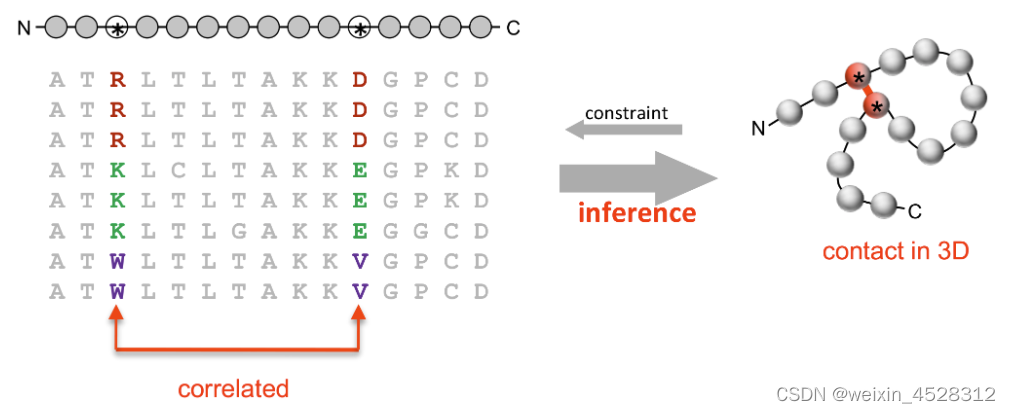
图1. 现有蛋白质的多序列比对可以抽取基因历史的共进化信息以及蛋白质残基间的空间结构关系。
蛋白质互作形成的复合物的MSA可以通过“合理”拼接蛋白质单体的MSA获得(图2)。但是,构造有效合理的蛋白质互作MSA需要满足一定条件。首先,单体MSA中的同源序列必须要到达一定的数目,以保证推导单体内部有效共进化信息的统计量。第二也更重要的是,蛋白质互作MSA需要提供蛋白质-蛋白质之间的空间演化信息。具体来讲,一个单体MSA中的同源序列与另一个单体的同源序列之间必须是来自于同一个复合体才能配对。否则,来自非相互作用或彼此相互独立的同源序列之间不存在相互作用的共进化关系,它们的错误配对会对整体的互作MSA以及最后的蛋白质互作建模产生负面的影响。因为目前缺少大标度高质量的蛋白质互作序列数据库供互作MSA的直接搜索,如何从单体MSA构造有效的互作MSA、进而产生高精度蛋白质复合物结构预测仍然是一个重大挑战。

图2. “合理”拼接单体MSA可以构建复合物MSA进而预测蛋白质复合物结构。
在这项研究中,张阳教授团队基于他们此前开发的DeepMSA迭代MSA构建算法,提出了改进版的DeepMSA2算法。DeepMSA2结合了一系列基于大标度宏基因组序列数据库的MSA生成策略,同时介绍了一种新的AI驱动的MSA打分策略来筛选高质量的MSA模型。定量的基准和双盲测试结果都表明,使用了DeepMSA2的结构预测算法DMFold可以构建远超AlphaFold2精度的蛋白质-蛋白质相互作用结构预测。
II. 原理设计
DeepMSA2算法包括DeepMSA2-Monomer单体MSA构建算法和DeepMSA2-Multimer复合物MSA构建算法。
对于蛋白质单体MSA构建,DeepMSA2-Monomer利用三个基于不同搜索策略构建的并行模块(dMSA、qMSA 和 mMSA)从不同的数据库中获取原始MSA。这些搜索策略都是基于动态规划(dynamic programming)和隐马尔科夫模型(hidden Markov model)算法,而数据库则由多个全基因组和宏基因组序列库组装而成。三个MSA生成模块都遵循类似的逻辑:首先在序列数据库检索初始的查询序列,如果未获得足够的有效序列,则对更大的数据库进行迭代搜索。最后利用快速AI蛋白质结构置信系数对三个模块收集到的原始MSA进行打分排序,选择出最佳MSA(图3)。

图3. DeepMSA2-Monomer构建蛋白质单体MSA的流程图。
对于蛋白质复互作合物MSA 构建,DeepMSA2-Multimer首先从每条单链数据中选取M个单体MSA,与其它所有N个单链的前M个单体MSA一一配对,从而得到M*N个联合MSA。然后,每个联合MSA中的序列通过连接来自不同组分单链的、具有相同物种起源的蛋白质单体序列来创建复合物的MSA。最后,DeepMSA2根据联合MSA的深度以及单体MSA的AI建模置信系数的综合得分来选择最佳复合物MSA(图4)。

图4. DeepMSA2-Multimer构建蛋白质互作MSA的流程图。
最后,通过将DeepMSA2算法与AlphaFold2端到端结构训练模块的结合,该研究提出了蛋白质单体及复合物三维结构预测算法DMFold。DMFold算法也包括两个部分,蛋白质单体结构预测算法DMFold-Monomer(简称DMFold)和蛋白质复合物结构预测算法DMFold-Multimer。
III. 结果分析
(1) 蛋白质单体结构预测
这项研究首先在蛋白质单体预测问题上与AlphaFold2算法进行了比较。AlphaFold2是谷歌DeepMind团队开发的第二款蛋白质结构预测软件,代表了该领域目前最为先进的AI预测算法。图5展示了DMFold与AlphaFold2在 132个非冗余的困难蛋白质单体上的TM-score比较。TM-score是由张阳实验室首先提出、后来被业界广泛使用的一种用来评估蛋白质结构预测准确与否的指标。TM-score=0代表最差预测,TM-score=1代表预测的和真实的结构一模一样,而TM-score=0.5表示预测的模型和真实结构有着大致相同的拓扑学结构。总的来说,DMFold预测模型的TM-score比AlphaFold2高出5%(0.82 vs 0.78)。特别地,在图5红色框内所示的特别困难蛋白上,DMFold的精度要显著地优于AlphaFold2(0.63 vs 0.52)。
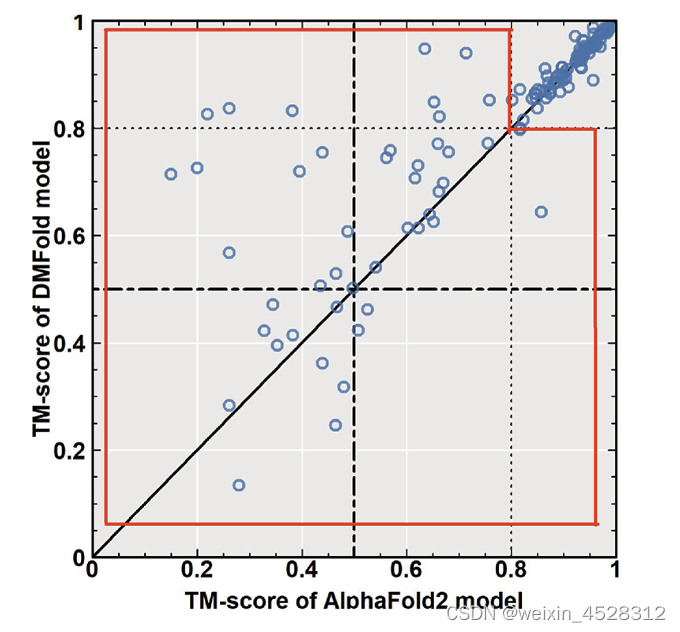
图5. DMFold与AlphaFold2对蛋白质单体结构的预测精度的比较。
图6以一种RNA聚合酶(PDB ID: 6vr4)作为示例,比较了DMFold与AlphaFold2的结构预测精度。首先,AlphaFold2预测出的结构模型的TM-score为0.20,全局pLDDT分数为0.40。而DMFold结构模型的TM-score为0.73,全局pLDDT分数为0.71。pLDDT分数是AlphaFold2用来评估结构预测质量的置信系数,其中,pLDDT≥0.7表示预测的结构可能正确,pLDDT<0.7表示预测的结构可能错误。从图6展示的结果来看,DMFold预测出的结构模型可以得到更高的TM-score精度以及pLDDT置信系数。

图6. DMFold和AlphaFold2对RNA聚合酶的预测结构精度的比较。
为了验证该算法对于大规模结构建模的实用性,作者进一步将DMFold应用于人类蛋白质组的结构预测。他们选取了AlphaFold2 DB数据库中5042个AlphaFold2预测失败(pLDDT<0.7)的人类蛋白。根据DMFold和AlphaFold2 DB数据库对这5042个蛋白质的pLDDT分数直方图分布显示,DMFold能够成功预测其中1934个人类蛋白的结构(图7A)。对这1934个人类蛋白的残基水平pLDDT分数的进一步比较显示,DMFold预测的氨基酸水平置信系数在93%的情况下都比相应的AlphaFold2的结果更高(图7B)。

图7. DMFold和AlphaFold2对5042个人类困难蛋白质的结构预测结果比较。
(2) 蛋白质-蛋白质复合物结构预测
为了测试DMFold对蛋白质互作的结构预测能力,该研究从早期CASP比赛(即CASP13和CASP14)中,收集了54个蛋白质复合物。这些复合物包括了2至8个单链蛋白,其中有14个是异源复合物,40个是同源复合物。图8比较了DMFold-Multimer与AlphaFold2-Multimer对这54蛋白复合的结构预测精度。平均来讲,DMFold-Multimer预测的模型的TM-score为0.83,而AlphaFold2-Multimer的精度为0.74。

图8. DMFold-Multimer与AlphaFold2-Multimer对蛋白质复合物结构预测精度的比较。
分析显示,DMFold-Multimer之所以能够产生更高质量蛋白质互作结构模型的原因主要在于两方面:第一,DeepMSA2-Multimer提出了一种全新的MSA构建、排序、配对和选择机制。第二,DeepMSA2-Multimer收集创建了一个包含约400亿个蛋白质序列的宏基因组数据库。因此,DeepMSA2-Multimer能够构造更加精准反映蛋白质互作共进化的MSA。相应地,DMFold-Multimer能够在两个方面提升蛋白质复合物的三维结构模型精度:一是提升组成蛋白质复合物的单体蛋白的预测精度(图9,T0988o);二是提升单体蛋白间的相对方位(包含蛋白质接触界面)的预测精度(图9,T1038o)。这些结果表明互作MSA的正确构建对于蛋白质复合物结构预测至关重要。
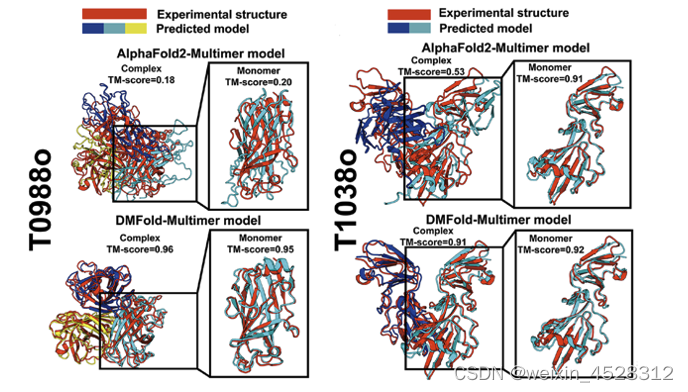
图9. DMFold-Multimer与AlphaFold2-Multimer对两个典型蛋白质复合物(T0988o,T1038o)的结构预测精度比较。
(3) CASP大赛盲测结果
DMFold参加了2022年举行的最近一期蛋白质结构预测大赛(CASP15,参赛名:Zheng),并在蛋白质-蛋白质复合物预测比赛中排名第一。CASP是世界范围的蛋白质结构预测比赛,每两年举行一次,旨在对领域内的蛋白质结构预测技术做出客观的测试和评价。CASP采用严格的双盲预测机制,被誉为评估蛋白质结构预测技术的金标准,也被业界视为“蛋白质结构预测的奥林匹克”。张阳团队在过去16年举办的八届CASP大赛中(CASP7-14),均取得了优异的成绩,连续八次取得自动组蛋白质结构预测冠军。
蛋白质-蛋白质相互作用结构预测是CASP近年来引入的新赛道。按照CASP15组委会对蛋白质互作结构预测给出的官方Z-score打分结果,DMFold-Multimer整体分数(35.4)比标准版的AlphaFold2-Multimer(12.3)高出2.9倍;同时也比所以参赛团队的第二名(29.9)和第三名(28.4)分别高出18%和25%(图10)。

图10. CASP15世界蛋白质结构预测大赛蛋白质-蛋白质复合物预测的官方排名结果。
图11进一步展示了DMFold-Multimer算法在CASP15中对27个蛋白质复合物构建的双盲预测结构模型。这些复合物结构模型的TM-score均大于0.8。其中,7个复合物(H1111、H1114、H1137、T1170o、H1171、H1172和T1181o)具有超大尺寸,分别包含8460、7988、4592、1908、1956、2004和2064个氨基酸残基,而DMFold-Multimer预测模型的TM-score分别为0.98、0.91、0.94、0.93、0.93、0.91和0.85。最为复杂的复合物是H1137,其化学计量式为“A1B1C1D1E1F1G2H1I1”,包含9种不同的蛋白质的10条子链,而DMFold-Multimer对该超大复合物结构预测的TM-score为0.94。长期以来,对大尺寸蛋白质复合物的结构预测被认为是分子生物学的一个重大挑战。这些结果表明,DMFold-Multimer算法能够对许多超大尺寸蛋白质-蛋白质复合物结构进行精准预测。这一成功标志着该领域朝向最终解决蛋白质互作结构模建问题迈出了重要的一步。
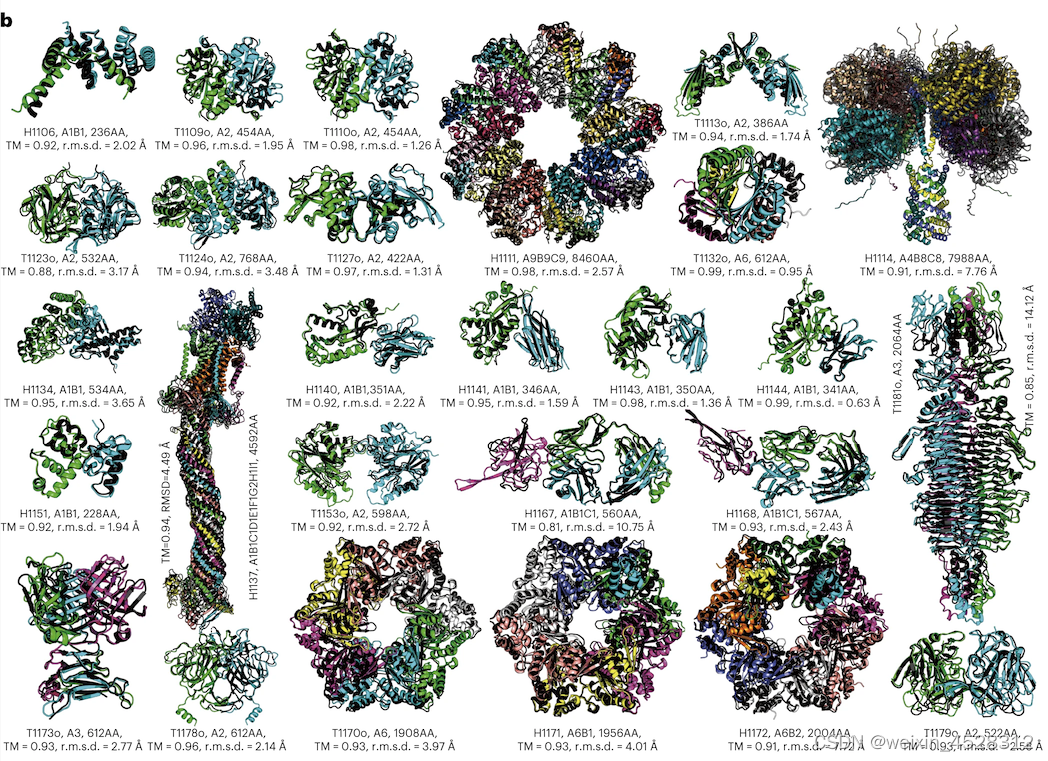
图11. DMFold-Multimer在27个蛋白质互作复合物上预测的结构模型(彩色)与实验结构(黑色)的重叠比较。
抗体-抗原蛋白的相互作用对人体抵御外界病原体的入侵至关重要。一般的普通抗体包括两个重链和两个轻链,形成复杂的Y型结构。而纳米抗体是一种只含有一个可变结构域的特殊抗体,它通过与特定的抗原相互作用启动关键的免疫反应。图12展示了DMFold-Multimer和AlphaFold2-Multimer对CASP15中三个纳米抗体-抗原复合物(H1140、H1141和H1144)的结构预测结果。这三个复合物的抗原均为小鼠的CNPase,而抗体则是三种不同的纳米抗体。AlphaFold2-Multimer算法预测的复合物模型的TM-score相对较低,均低于0.7。相比之下,DMFold-Multimer表现出了卓越的预测能力,其所构建的三个结构模型的TM-score分别为0.92、0.95和0.99,几乎可以媲美实验解析的结构精度。

图12. CASP15比赛中纳米抗体-抗原复合物的结构预测案例分析。
IV. 结论与展望
这项研究开发了两款基于AI的蛋白质相互作用结构预测软件。其中DeepMSA2可用于从宏基因序列数据库中搜索构建多序列比对和蛋白质互作共进化信息,而DMFold则用于从MSA和共进化数据中构造蛋白质复合物三维结构模型。
实验表明,DeepMSA2算法可以显著提高蛋白质复合物结构预测的准确性。与现有的MSA构建方法相比,DeepMSA2的一大优势在于快速迭代搜索和基于AI的模型预选策略。前者使得该算法可以检索海量的宏基因组序列数据库,有助于增加所得MSA的多样性和覆盖范围,而后者通过AI训练筛选出能够更加精确的反映共进化信息的MSA,进而提升基于AI的蛋白质结构预测的精度。
虽然DeepMSA2/DMFold的最初目标是针对蛋白质-蛋白质复合物四级结构的构建,但是它们也可以同时用于对蛋白质单体三级结构的预测。大规模的基准和双盲测试都表明,DeepMSA2/DMFold对蛋白质单体和复合物的结构预测都可以产生远超目前标准版本的AlphaFold2的预测精度。
尽管如此,DeepMSA2/DMFold仍然存在一些挑战。例如,DeepMSA2的复合物MSA是从单体MSA联合构建而来的。因此亟需解决的一个基本问题是如何有效连接不同组分MSA的序列以形成最佳复合物MSA。目前基于物种注释的序列连接机制仅适用于基因组序列,无法充分利用宏基因组数据库中信息丰富的同源序列来指导复合体结构组装。如何优化基于宏基因组的MSA配对与构建,对于进一步改进蛋白质互作结构预测精度以及相关研究(例如预测任意两个蛋白质是否为相互作用)都具有重大意义。

新加坡国立大学和密歇根大学张阳教授
参考文献:
Zheng, W., Wuyun, Q., Li, Y. et al. Improving deep learning protein monomer and complex structure prediction using DeepMSA2 with huge metagenomics data. Nat Methods (2024). https://doi.org/10.1038/s41592-023-02130-4
- 先引入
赞
踩

