
- 1【Android学习】自定义文本框和输入监听
- 2Android Material Design之CardView(卡片式布局)_android materialcardview
- 3必看干货 | 学习Vivado如何获取License_vivado里面如何更换许可证
- 4成功解决,电脑重置后出现不支持标准硬件安全性,页面不可用等_设备安全性转到设置页面不可用怎么办
- 5Ant内置任务之sql_ant 执行sql语句
- 6程序员用Python赚外快的具体指南!找点轻松的活增加收入_python做外包快吗
- 7LINUX——账号和权限管理_linux 设置账号权限
- 8sql大数据 基础(DCL 数据控制语言)_大数据应用基础sql语音
- 9确实会画PCB会看原理图之后我再看嵌入式项目,整个系统得多_嵌入式需要画pcb和原理图吗
- 10脂质体优良材料DSPE-PEG-COOH二硬脂酰基磷脂酰乙醇胺-聚乙二醇-羧基---为华生物_dspe-peg-nh2胶束 酸碱度
Biological Psychiatry:人脑转录组成像的最佳实践
赞
踩
导读
现代全脑转录图谱为研究大脑组织的分子相关性提供了前所未有的机会,这些分子相关性通过非侵入性神经成像进行量化分析。然而,将神经成像数据与转录组测量相结合并非易事,需要仔细考虑才能做出有效的推断。本文回顾了最近的研究工作,探索了不同方法选择如何影响成像转录组分析的三个主要阶段,包括①转录图谱数据的处理;②将转录测量与独立衍生的神经影像学表型联系起来;③通过基因富集分析评估已识别关联的功能意义。本研究的目标是促进这一快速发展的领域开发标准化和可重复的方法。本文确定了方法可变性的来源、可能影响研究结果的关键选择,以及减少假阳性或虚假结果的考虑因素。最后,本文提供了在所有三个分析阶段中实现当前最佳实践过程的免费开源工具箱和人脑转录组数据库资源。
前言
精神疾病通常表现为大脑连接障碍。磁共振成像为研究人员提供了一种强大的工具,用于绘制一系列不同疾病中大脑连接障碍的各个方面,但神经成像本身无法揭示这些中断的潜在分子机制。一个富有成效的方法是研究神经成像衍生的连接组表型的遗传基础。随着大脑发育,分子梯度在引导轴突到达其靶点方面起着关键作用,而大脑结构和功能的许多方面,包括大脑连接,都显示出一定程度的遗传性。因此,确定与精神疾病有关的连接组表型的遗传驱动因素可能为了解支撑大脑组织的复杂分子机制提供了一扇窗。
在这种情况下,影像遗传学的主要方法涉及通过遗传关联研究将某些连接组或其他表型的表型变体与DNA的结构变体联系起来。虽然这种方法可用于识别与表型相关的单核苷酸多态性,但不能保证相关变体在驱动表型变异性中起因果作用,因为它可能与实际的因果变体存在连锁不平衡状态。在许多情况下,如果不进一步研究,可能也很难知道变体对基因表达的分子效应。虽然直接分析转录活性的研究可能会揭示与病理生理学更密切相关的分子过程,但它们历来仅应用于相对较小的组织块,因此很难将这些测量与神经影像学表型联系起来。
过去十年见证了高通量组织处理和分析的重大进展,使生成解剖学上全面的大脑转录组图谱成为可能。这些图谱包含了几乎整个基因组转录活性的测量数据,这些测量数据来自大脑中数千个组织样本,为识别具有与神经影像学表型相关的空间表达模式的基因开辟了新的可能性,从而催生了新兴的成像转录组学领域。这些分析已经确定了大脑多种结构和功能特性的转录相关性,包括以下方面:区域间连接;经典的sensory-fugal皮质层次;典型大规模网络及其拓扑和时间特性;以及内在动力学的空间梯度。成像转录组学也确定了不同疾病中大脑连接障碍的转录相关因素,并揭示了在疾病以及发育和衰老过程中观察到的大脑变化的假定细胞相关因素。此外,转录数据为脑功能的动态模型提供了信息,并提高了我们对神经退行性疾病传播的理解。
尽管成像转录组学前景广阔且迅速普及,但神经成像与全脑转录组-图谱数据的整合取决于众多可能影响最终结果的数据处理和分析选择。不同的研究人员历来都使用自己定制的分析管道,因此很难确定这些选择对报告结果的影响。因此,最佳实践工作流的开发对于以有效和可重复的方式推进该领域至关重要。本文回顾了近期探索各种处理选项影响的工作,并试图开发易于使用的管道以实现最佳处理选择,从而构建成像转录组学的最佳实践工作流。本文概述了此类分析中的一些关键步骤,强调了需要仔细考虑的问题,并在已有基础上给出了最佳选择的建议。本文将重点讨论任何成像转录组分析的3个关键阶段:1)处理转录图谱数据;2)将表达测量与神经影像学表型相关联;3)评估基因特异性和富集性。本文希望促进该领域标准化处理和分析方法的发展,从而促进不同研究之间的比较,并促进有效和准确的推断。
阶段1:处理转录图谱数据
成像转录组学研究依赖于全脑转录图谱,可量化大脑中多个位置数千个基因的表达。测量转录活性的方法取决于一系列因素,包括物种、所需的空间分辨率和组织可用性。与其他具有更高空间分辨率的方法(如单细胞RNA测序或原位杂交)相比,人脑组织的有限可用性意味着大块组织微阵列仍然是高通量空间转录组学最容易获得的方法。艾伦人脑图谱(AHBA)提供了一个解剖学上全面的人脑转录图谱,对来自6个死后大脑的3702个解剖位置的20000多个基因的表达进行了量化分析。由于使用T1加权磁共振成像对供体大脑进行扫描,并将扫描结果归一化到标准化空间,因此可以直接比较基因表达和神经成像数据的空间模式。然而,由于AHBA中的解剖位置是使用空间分布的小块组织进行采样,因此实现此空间映射的最简单方法是对两者应用一些区域分割。通过这种方式,特定脑区内基因表达的汇总测量可以与同一区域内量化的成像测量相关联。
类AHBA数据中的主要任务是构建一个N×G矩阵,其中N是分割的脑区数量,G是测定的基因数量。图1概述了获取该区域基因表达矩阵的典型工作流程的关键步骤。每个步骤都需要研究人员做出可能影响最终结果的选择。在这里,本文总结了每个步骤的关键点和建议。
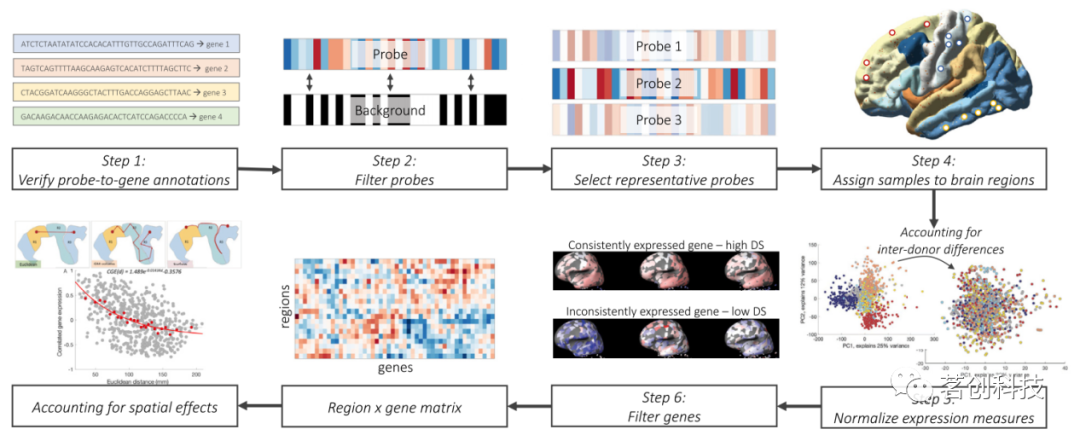
图1.艾伦人脑图谱处理步骤示意图。
步骤1:验证探针到基因的注释
微阵列数据使用探针序列来量化基因表达,探针序列对应于包含单个基因的DNA的独特部分。基因探针的分配是使用不断更新的可用测序数据库来进行的。因此,使用最新、最准确的基因探针定位来确保测量的有效性是至关重要的。
步骤2:过滤探针
微阵列实验容易产生背景噪声,部分原因是非特异性杂交。因此,必须去除表达水平不超过背景的噪声探针,以提高微阵列测量的有效性。根据本研究的分析,在至少50%的样本中保留表达水平超过背景水平的探针可以为降低噪声提供适当的平衡,同时保持大约70%的可用探针,从而确保高解剖覆盖率。
步骤3:选择有代表性的探针
原始AHBA数据中90%以上的基因具有多个探针可用,而且并非所有探针都显示出一致的表达模式。应选择单个有代表性的探针或跨多个探针的汇总测量值,以降低数据复杂性和帮助解释。已有文献实现了多种方法。本文建议根据探针与2个(共6个)供体大脑中收集的RNA测序测量值的相关性来选择探针,因为它提供了最接近地面真值测量的方法。当无法做到这一点时,本文建议使用具有最高差异稳定性的探针,用于测量供体大脑中基因表达谱的一致性。
步骤4:将组织样本分配到脑区
每个组织样本都具有立体定向坐标和解剖标记的特征,可用于将测量到的相应表达映射到所选大脑分割中的一个区域。本文建议根据组织样本的广泛解剖位置(皮层/皮层下)将其分别映射到区域,并将样本分配到分割中最近的区域(而不是该区域的质心),同时将原始样本位置的距离阈值设置为2mm,以避免距离该区域太远的样本映射不准确。
步骤5:规范表达测量
由于AHBA数据是从6个供体大脑中收集的,因此任何结合不同大脑样本以获得解剖学全面图谱的分析都必须考虑供体大脑表达的个体差异。AHBA团队在数据发布之前执行的标准化程序消除了批次效应和人为的个体间差异,但仍残存大量的个体间差异。在每个供体脑中跨区域执行额外的z评分或缩放的鲁棒sigmoid归一化步骤可用于消除这种残余变异性,并最大限度地减少离群值的影响。
步骤6:在供体大脑中选择具有一致表达模式的基因
在AHBA的20000多个基因中,只有一小部分在不同的供体大脑中表现出一致的区域差异,这是通过差异稳定性测量进行量化的。识别在大脑中显示出可重复变异的一致表达基因或已知在大脑中表达的基因,可以为研究与神经影像学表型的关系提供更有针对性的方法。
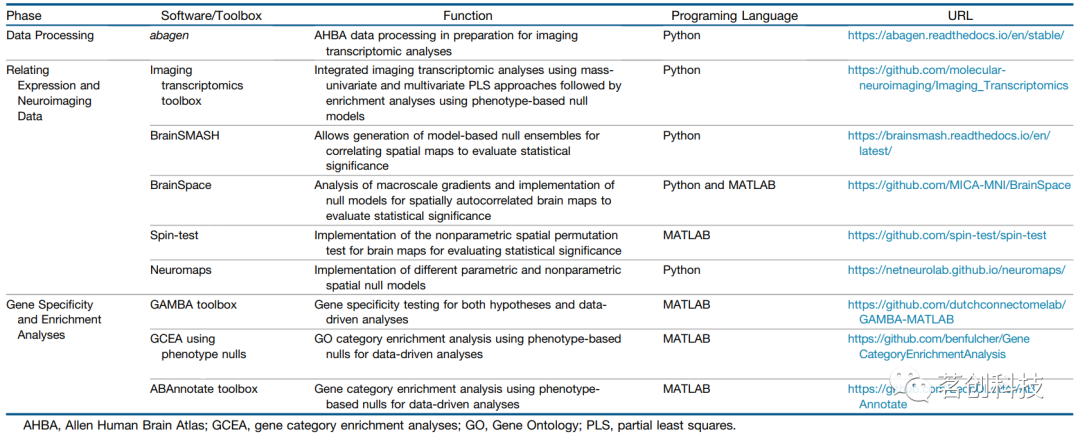
在以上概述的6个步骤中,每个步骤都可以做出大量的选择,从而导致研究人员可以选择的可能管道组合激增。最近的一项研究广泛探索了以上工作流中17个决策点的影响,产生了746496个不同的处理管道(见图2)。该研究使用这种综合分析来确定影响最终结果的关键处理选择,并使用基于3种常用分析类型的3个结果指标进行量化:基因共表达(跨区域基因之间的相似性),区域基因表达(特定基因或基因集的空间表达模式)和相关基因表达(CGE)(跨基因区域表达谱之间的相似性;下面将详细讨论所有测量;参见图3)。该研究发现,涉及基因归一化的数据处理选择(步骤5)影响最大(图2),其次是将组织样本映射到大脑区域的选择(步骤4)。与探针选择(步骤3)有关的影响最小(图2A)。通过将这项分析的结果与先前的结果相结合,该研究开发了一个在开源abagen工具箱中执行的数据处理工作流。abagen工具箱还可以生成所选处理选项的标准化报告,以提高出版物报告的透明度。
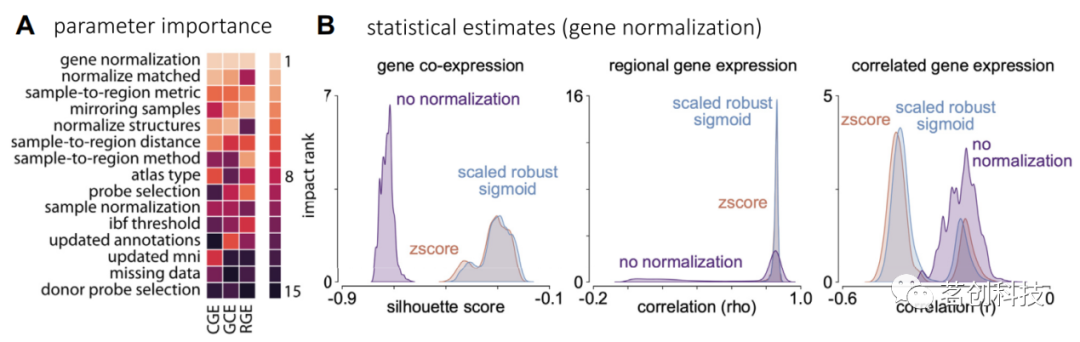
图2.数据处理选项的影响。
阶段2:相关表达和神经成像测量
将转录数据转换成区域-基因表达矩阵后,下一步是将这些测量与某些神经影像学表型相关联。在这种情况下,通常使用三种主要方法之一来概括基因表达数据(图3)。第一种是基因共表达分析,包括分析成对基因(跨脑区)表达模式之间的空间相关性。对于所有基因对,结果可以表示为一个(对称的)基因-基因矩阵,以分析基因表达相似性的模式(图3A)。该矩阵也可以使用一些聚合值(例如,矩阵或其子分量的特征向量,有时称为特征基因)进行概括,从而产生一个高度解释性的分量作为可以与成像数据相关联的空间地图。第二种分析侧重于区域基因表达,其中根据每个脑区定义的神经影像学指标评估选定基因或基因组的空间相关性。同样的原则适用于多变量分析,例如那些使用偏最小二乘法的分析,这些分析可以确定具有最大协方差的基因和成像测量的加权组合(图3B)。第三种分析考察了CGE,计算了所有成对脑区之间的相关性,量化了其基因表达谱的相似性。结果可以表示为一个(对称的)区域-区域矩阵,并直接与在区域对水平上测量的其他类型的数据进行比较,例如结构或功能连接(图3C)。
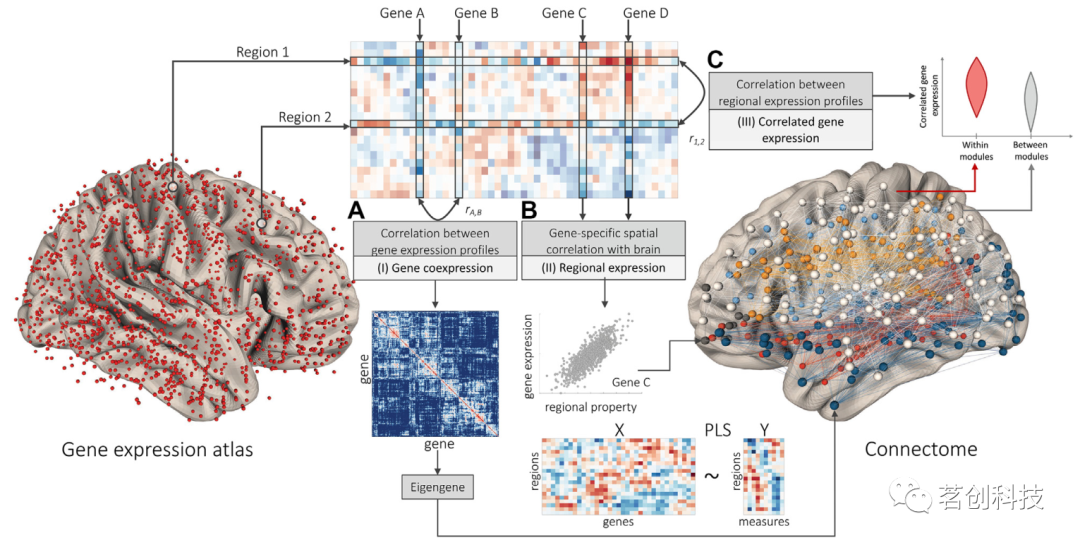
图3.成像转录组分析的类型。
大多数研究依赖于区域基因表达分析或CGE估计,并使用空间(大规模)单变量、全连接组或多变量分析技术将其与神经影像学测量相关联。分析方法之间的选择取决于正在研究的特定假设和影像学表型。例如,神经影像学表型的区域差异可以使用数据驱动和假设驱动的方法与基因表达的区域模式相关,而来自结构或功能数据的成对大脑连接的测量可能更自然地与CGE相关联,CGE可以捕获大脑区域之间共享的转录模式。转录和神经成像数据的一个重要特性是强烈的空间自相关;距离较近的区域比距离较远的区域具有更相似的数值。在基因表达数据中,任意两点之间的相关性随着它们的空间分离大致呈指数衰减。CGE中的这种指数距离规则已在不同类型的空间分辨转录组数据中得到确认,包括人类皮层,成年小鼠大脑,以及秀丽隐杆线虫的头部。对于动物的连接概率和强度以及人脑的区域间结构连接,已确定了类似的指数距离规则。
由于传统的统计方法假设观测值是独立的,因此分析空间自相关(即非独立)数据需要特别考虑,因为自相关可能会夸大表达和成像数据之间的关联。因此,如果没有考虑空间非独立性,可能会对真实关联产生过于乐观的估计。解决空间自相关的一种方法是建模并消除数据的空间依赖性(例如,通过回归),然后分析残差。这种方法非常适合成对区域特性(如CGE)的分析,但它基于以下假设:建模的空间相关性能够很好地近似于必须去除的空间自相关性。
另一种非常适合区域分析的方法是使用空间约束的零模型。这些零模型保留了空间映射的自相关性,从而能够推断经验观察到的表达和神经成像测量之间的相关性是否超出2个随机自相关变量的预期(图4)。非参数空间排列模型和参数化空间模型是神经成像研究中两大类受空间约束的模型。非参数模型非常适合于大脑皮层的分析,并利用皮层表面可以映射到球体的事实,允许简单的旋转,从而将值分配到特定的皮层位置,同时保留数据的确切距离依赖性。参数化模型估计经验地图的内在空间自相关性,并使用所得模型生成具有随机拓扑但具有空间自相关性的地形图。

图4.空间零模型。
空间排列模型的主要优点是保留了经验数据的距离-依赖关系。但是,含缺失值的区域可能会在地形图旋转后产生问题,并且已经开发了各种启发式方法来解决此问题,例如丢弃缺失数据,然后基于最近的可用区域对缺失值区域进行插值。另一个限制是,基于排列的方法不能应用于皮层下结构,这些结构通常不能充分建模为二维球面。参数化模型不受缺失数据的影响,可以等效地应用于皮层和皮层下测量,但不能保证经验距离依赖性与原始经验值的精确匹配。因此,涉及不同空间地形图的分析可能会受到模型拟合变化的影响。对这些不同皮层分析方法的各种实现进行比较表明,与参数化模型相比,基于排列的模型提供了更保守的显著性估计值和更低的错误率。然而,没有一种方法是完美的,对于强自相关数据的错误率超过40%,这表明有效的推断还需要进一步发展。Markello等人(2021)对不同的零模型进行了扩展研究,其实现工具箱可在https://netneurolab.github.io/neuromaps/上找到。
阶段3:评估基因特异性和富集性
目前全脑转录图谱量化了多达约20000个基因的表达水平。鉴于一组给定的影像学表型可能产生大量的关联,确定某些基因或基因集是否与感兴趣的表型优先相关至关重要。分析可以大致分为以下两类:1)假设驱动,即分析假设感兴趣的特定基因(或基因集);2)数据驱动,即计算整个转录组的效应,然后推断特定基因[或功能分类的基因集]的优先参与。
假设驱动分析已被证明有助于理解不同疾病中假定易感基因的表达谱与大脑变化之间的联系,以及某些特定基因的表达与连接相关表型之间的联系。细胞特异性标记基因的转录谱显示与一系列疾病的皮层厚度变化以及与年龄相关的皮层髓鞘化和厚度变化有关。大部分的研究工作都忽略了这样一个问题,即所报告的关联是特定于所选择的基因集,还是可以使用不同的基因集来识别相似的关联。考虑到基因表达数据的低维性,其中常见的大规模梯度可以解释基因间转录变异的很大一部分,通过比较表达和神经成像测量之间已识别的关联与使用其他基因集观察到的影响来评估基因特异性至关重要。实证研究结果表明,即使在使用空间约束零模型评估的空间特异性关联中,使用随机基因集的自由基因特异性测试中仅58%存活,使用随机选择的大脑中特异性表达的基因时仅37%存活。模拟进一步表明,在空间自相关脑表型和单基因转录谱之间识别出的所有关联中,只有3%在空间自相关和基因特异性校正后幸存。这一结果表明,对空间自相关性的控制不足以识别特定的基因-表型关联,而且文献中大量看似有意义的关联可能并不特定于所报告的基因集。因此,在选择一个适当匹配的基因集来评估研究结果的特异性时,应仔细考虑对数据提出的具体问题。考虑到大量相关基因可能会掩盖特异性或主导结果,尤其是在多变量分析中,首先检查感兴趣基因与未包含在目标集中的任何其他基因之间的空间相关性始终是一种良好的实践。
与假设驱动的研究相比,数据驱动的分析测试的是在转录数据中分析的数千个基因的表达谱与感兴趣的表型之间的关联。然而,许多基因并不是独立的,它们具有相似的空间相关表达模式或作为共同生理通路的一部分共同工作。因此,在数据驱动的分析中,测试与表型特别强的关联是否集中在某些功能相关的基因集中是很常见的。这些分析中最常见的方法是使用基于层次系统的基因功能注释,根据基因与分子功能、细胞组分和生物过程的关联对基因进行分类。传统上,这种基因类别富集分析(GCEA)已被用于支持全基因集关联结果的解释,或者用于对特定组织样本的基因表达进行病例对照比较,通过评估与随机选择的基因相比,某一基因类别是否优先与表型相关(图5A)。
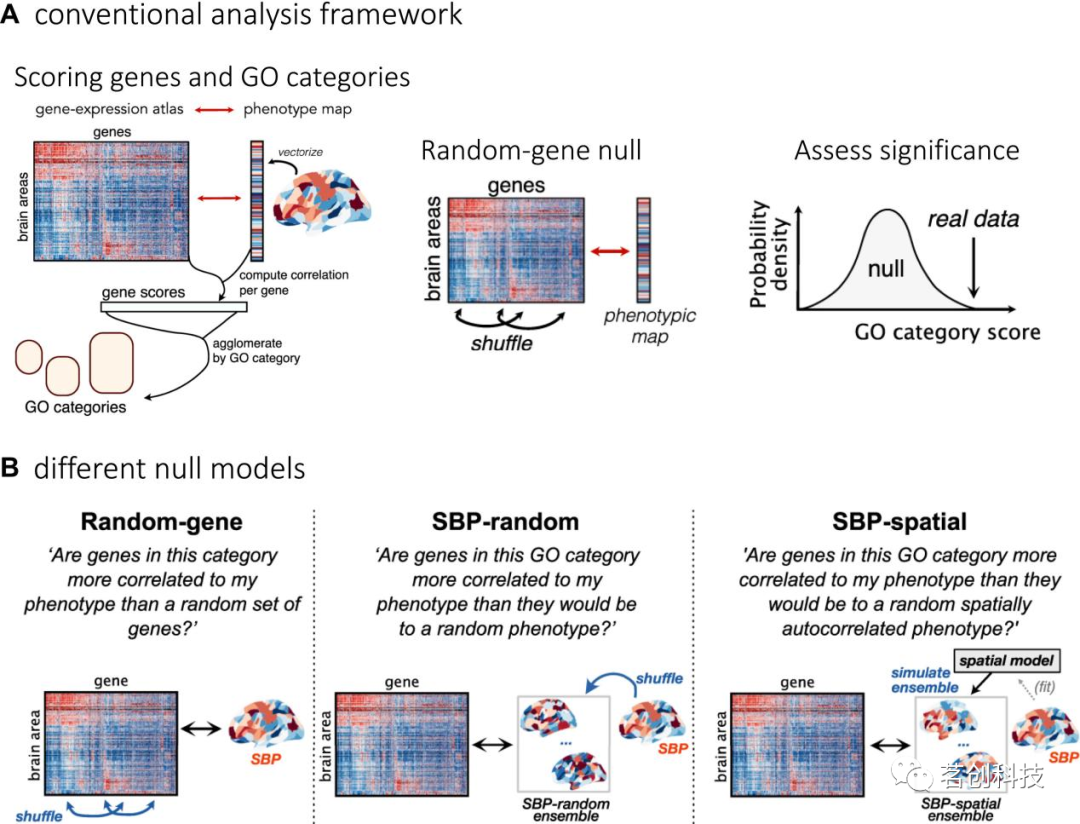
图5.基因类别富集分析。
将GCEAs应用于空间嵌入的转录数据会引入额外的统计考虑因素,这些考虑因素可能导致虚假的富集证据。用于GCEA的经典零模型没有考虑这种相互关联结构(或空间自相关),导致统计推断趋向于假阳性(图5A)。Fulcher等人(2021)建议通过随机化表型(而非基因)来生成零分布,从而在零样本的类别中保留基因-基因相关结构。从上一节可以明显看出,表型可以以空间约束或非约束的方式随机化(图5B)。使用这种以表型为中心的零模型进行推断,大大减少了在对小鼠和人类的14种不同脑表型进行分析时确定的显著富集结果的数量。这些发现表明,文献中报道的大量富集结果可能受到假阳性偏倚的影响,需要进一步的研究来验证。
结论
本文概述了与进行成像转录组学分析相关的几个关键考虑因素(图6)。对这些考虑因素的详细研究强调了在做出处理和分析选择时需要注意的事项。目前已经开发了几个开源工具箱,可以实现每个分析阶段列出的许多最佳实践过程(表1)。然而,即使依赖于这些工具,也需要根据具体情况仔细考虑,以获得有效的推断和可再现的结果。此外,任何使用AHBA数据的分析都必须考虑几个关键的限制因素。

图6.使用艾伦人脑图谱在3个分析阶段对人脑进行转录组成像分析的一般建议。
表1.每个分析阶段可用的工具箱,包括数据处理、评估基因表达和脑成像测量之间的关联,以及通过基因富集分析评估这些关联的特异性。
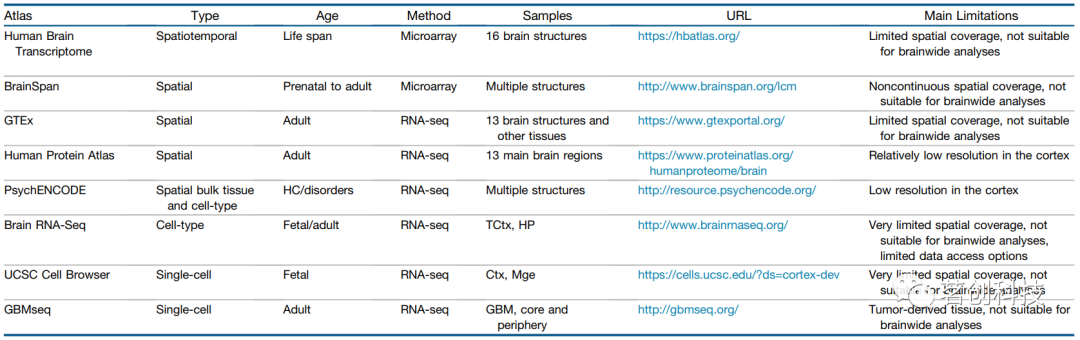
首先,基因表达与蛋白质丰度之间的关系很复杂,转录活性的变化不一定会影响蛋白质水平。其次,AHBA依赖于对大量组织样本的微阵列分析,由此产生的估计值可能受到细胞组成的区域差异的影响;因此,应尽可能使用单细胞RNA测序数据对结果进行交叉验证。第三,虽然AHBA旨在量化典型的转录模式,这些模式与其他死后组织库(表2)相比提供了无与伦比的空间覆盖率,但AHBA测量来自6个成年供体的小样本。AHBA各脑区基因表达的变异性比这6个个体大得多,这提示该图谱可用于研究稳健的区域表达谱,但对供体大脑代表性的担忧是有原因的。第四,AHBA仅测定成人大脑中的基因表达模式,但许多神经表型可能取决于发育过程中复杂且动态变化的基因表达模式。最后,由于AHBA数据是基于小样本的供体测量,因此在基因表达模式与影像学表型(或其他结果)之间发现的任何关系都是纯粹的相关关系,并不直接揭示因果机制。
表2.人脑转录组数据库。
原文:Toward Best Practices for Imaging Transcriptomics of the Human Brain.

