
- 1循环神经网络 (处理时序型数据)_处理时序数据的神经网络
- 2filter多个参数处理方法+Python的filter过滤器理解_过滤器怎么处理参数
- 32023年西工大noj(C程序设计)100题代码参考_noj西工大
- 4Python中对CSV数据预处理_csv文件预处理
- 5自然语言处理入门(何晗):第一章_自然语言处理入门何晗电子版
- 6Git 1(git安装与基本使用、在idea里面使用gitee)_不用git客户端也可以使用git吗
- 7自然语言处理 | (13)kenLM统计语言模型构建与应用_lm arpa
- 8Visutal Studio2022 如何使用Github copilot_visual studio copilot
- 9空间注意力模块与通道注意力模块实现
- 10DiffusionCLIP: Text-Guided Diffusion Models for Robust Image Manipulation
文献阅读【RNA-seq数据归一化】_rna-seq条件分位数归一化
赞
踩
最近一直在做lncRNA的分析,其中的lncRNA的差异表达分析中,需要对reads count 进行归一化,之前没有考虑很多,就用的通常的流程:
hisat2→stringtie→prepDE.py/featureCount→DESeq2
其中的DESeq2 的归一化部分,也是我们通常称的标准化,是我们关注的重点,DESeq2主要原理:通过计算一个归一化因子,并进行变换,进而提高中等表达基因的地位。 归一化和标准化总是搞不清,我还特地查了二者的共性和区别:
共性:归一化和标准化本质上都是一种线性变换。线性变换保持线性组合与线性关系式不变,这保证了特定模型不会失效,归一化和标准化的本质都是缩放和平移。
区别:他们的区别直观的说就是归一化的缩放是 “拍扁” 统一到区间(0-1),而标准化的缩放是更加 “弹性” 和 “动态” 的,和整体样本的分布有很大的关系。
- 1
- 2
下文统一使用归一化。
这篇文献是我在查询归一化方法的时侯,Github上一个网友推荐《Comparing the normalization methods for the differential analysis of Illumina high-throughput RNA-Seq data》,翻译过来就是: 《Illumina 高通量 RNA-seq 数据差异分析的归一化方法比较 》。
下面开始介绍这篇2015年发表在 BMC Bioinformatics 文献。

如果觉得前面内容太多,可以直接跳到最后看总结。
一、背景和目的
1.1 背景:
RNA-seq 技术的快速发展和测序成本的降低使其成为一种广泛应用的基因表达定量技术。 由于归一化在RNA-seq 数据分析中的重要性,人们提出了各种归一化方法。 归一化方法: 非丰度估计)的归一化方法(non-abundance normalization 1. RC(row count):每个基因的原始计数是所有run基因计数的总和。 2. UQ(upper quartile):上四分位数是通过对所有样本的基因计数应用0.75的上四分位数来计算的,主要在芯片测序数据中使用。 3. Med(median):中位数计算为所有样本基因计数的中位数。 4. TMM(Trimmed mean of M-values normalization):M值的 trim 均值是一种用于RNA-seq 数据差异表达分析的标度归一化方法。 这种归一化方法是在R包 edgeR中实现的。 使用包中的 CalcNormFactors 函数计算缩放因子(scaling factors),然后通过将基因计数除以每次运行的每个缩放因子来获得重新缩放的基因计数。 TMM是所有样品 重新缩放(rescaled) 基因计数的总和。 5. DESeq:DESeq是一种基于负二项分布模型的差异基因表达分析方法, 方差和均值通过局部回归联系起来,并给出了一个也给出比例因子(scale factors) 的实现。 它在DESeq包中,通过 EstimateSizeFactorsFormatrix函数,可以计算每次运行的缩放因子。 将基因计数除以每个标度因子后,DESeq值被计算为所有样品重新缩放基因计数的总和。 6. Q (quantiles):分位数以前被用来归一化数组之间的单通道或A-value 芯片数据。 R包limma中的normalizequantiles函数将矩阵的列归一化为具有相同的分位数。 这里,我们将函数输出的总值设置为分位数的归一化值。 7. RPKM:这种方法通过对总转录本长度和测序 reads 数进行归一化,从RNA-seq数据中量化基因表达。 RPKM值可以使用以下定义轻松计算:  8. ERPKM::RPKM的变形体,采用effective transcript length,但是作用不大。由于reads的长度不为零,而reads 概率取决于有效长度,我们使用有效reads长度计算了每千贝每百万映射reads的有效转录本。
8. ERPKM::RPKM的变形体,采用effective transcript length,但是作用不大。由于reads的长度不为零,而reads 概率取决于有效长度,我们使用有效reads长度计算了每千贝每百万映射reads的有效转录本。 
丰度估计的归一化方法(abundance normalization):使用机器学习算法进行丰度估计
RSEM: 不同于以往的归一化方法。 提出了一种结合期望最大化算法的有向图模型来估计丰度。 RSEM提供了一个从RNA-SEQ数据中量化基因丰度的软件包,因此我们通过准备参考转录本数据和输入RNA-SEQ数据计算RSEM值来生成参考指数。
Sailfish:在丰度估计中,被介绍为无比对。它利用K-MER的概念对RNA-Seq reads进行索引和计数。 在这里,我们使用偏置后的估计K-MERS数作为估计计数。
- 1
- 2
- 3
1.2 目的:
对目前存在的归一化方法进行比较,以便为将来的实验选择最合适的方法产生合适的指导方针。
二、方法和材料
2.1 方法
==通俗解释:== 用 实验测量的 qRT-PCR值 和每种归一化计算出的RNA-seq的丰度(或者可以理解为归一化后的 reads count)预测进行比较, 用 可以评估 数据相关性 的统计方法,来评估每种归一化方法计算结果的准确性。
【这里我是存在疑惑的,qRT-PCR实验中也存在误差,其数据结果是否可行,重复了几次,文中作者仅使用原作者数据进行的评估,自己没有实际做实验得到 qRT-PCR 数据】
- 1
==学术解释:== 用Shapiro-Wilk正态性检验QRT-PCR值的分布和所有的归一化结果。 根据P值<0.05的检测结果,QRTPCR值和归一化结果均不呈正态分布。 为了对数据进行描述,我们使用Spearman的秩相关系数,通过计算每种归一化方法的RNA-Seq丰度预测与测量的QRT-PCR值之间的相似性来评估性能。
Spearman相关系数作为一种非参数方法来度量两个变量之间的线性相关性。 它被计算为数据秩上的皮尔逊相关系数。 对于一组大小为n的基因和相应的n个原始数据,变量 x 为 QRT-PCR 基因表达值,变量 y 为归一化方法的结果,相关Rs 使用下面的公式计算。
 Spearman相关系数将产生+1和-1之间的值,其中+1表示总的正相关,0表示不相关,而-1表示总的负相关。 最接近+1或-1的值表示最高的相关性,因此也是最好的归一化结果。
Spearman相关系数将产生+1和-1之间的值,其中+1表示总的正相关,0表示不相关,而-1表示总的负相关。 最接近+1或-1的值表示最高的相关性,因此也是最好的归一化结果。
2.2 材料
高通量 RNA-seq 数据收集自NCBI数据库中的SRA (Sequence Read Archive) 数据库,如果自己的实验室没有条件进行测序,又想做数据分析,可以在这个数据库下载数据,自己处理和分析。 脑组织(HBR)和组织类型混合物(UHR)的原始 RNA-seq 数据 35 bp reads length SRA010153.1 76 bp reads length SRA039286 一般现在的测序数据都是reads length 或者说插入片段长度都是 150或者151 bp的双端测序数据。
结果
3.1 归一化方法的比较
-
Spearman相关分析显示,无论reads 长度如何,RC,UQ,Med,TMM,DESeq 和Q 都没有明显改善基因表达正常化。 -
ERPKM并没有取得比RPKM更好的结果。 使用有效的转录本长度显然不能改善归一化结果
-
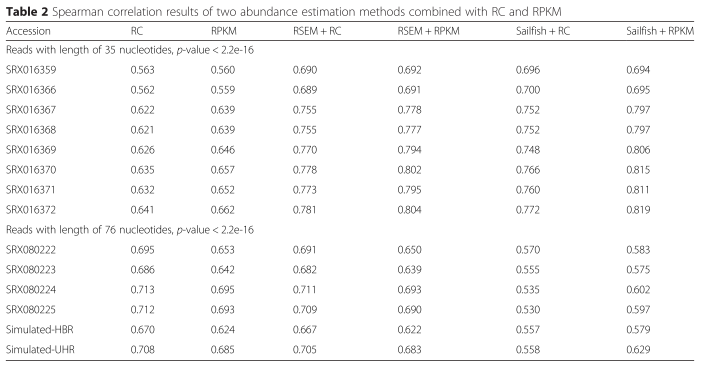
在实验结果中,RPKM结合Salifish 的归一化方法几乎可以取代qRT-PCR测量。 而对于76 bp 序列数据,采用无丰度估计归一化方法的RC获得了最好的结果,其次是相关性相似的RSEM; Salifish方法产生了更差的相关值。
-
从比较结果来看,归一化方法并不是所有序列数据都必须的。 TMM、DESeq和Q等样本间归一化方法无论 reads 长度如何都不能显著提高基因表达,但当比对精度较低时,RPKM可能更有效。
-
对于35 bp的reads 数据,在两种丰度估计归一化方法中,Salifish方法与RPKM结合的归一化结果比RSEM更好,因为它没有比对,也是一种相当高效的组合。 然而,==当比对精度较高时,RC似乎足够用于实际实验中的基因表达计算。 ==
-
表1给出了八种非丰度估计归一化方法(未应用丰度估计归一化)的Spearman相关系数结果。

-
表2给出了结合RC和RPKM的两种丰度估计方法(RSEM和Salifish方法)的Spearman相关系数结果。
3.2 添加 poly-A tails 的比较
-
换句话说,通过增加Poly-A尾长,可以将更多的 reads 映射到一个参考转录本。 -
总之,选择合适的Poly-A尾部长度可以改善差异分析; 然而,基于本研究中观察到的最小效应,Poly-A尾长可能可以忽略不计。
四、总结
-
对于reads长度为35 bp的样本,在8种非丰度估计归一化方法中,RPKM的相关值高于RC、UQ、MED、TMM和Q,证明在归一化中考虑转录本长度是相当有效的。
-
这个结果在我们进行短序列测序,例如miRNA测序的时侯就可以作为参考,一般的miRNA定量用的是RPM,华中农大夏瑞老师课题组开发的sRNAminer 中用的是 RP10M (reads per 10 million),也就是说,RPM主要应用于sRNA(sRNA长度变化不大),来消除测序深度bias;而RPKM/FPKM应用范围会更广泛,用于同时消除长度及测序深度这两种bias。
-
-
通过使用有效转录本长度,ERPKM与RPKM相比并没有改善归一化结果。 在结合丰度估计归一化方法后,对归一化结果进行了改进。 特别是RPKM和Salifish结合,我们建议研究者在未来的分析中作为一种归一化方法,几乎可以取代QRT-PCR,因为观察到了近0.8的相关性。 而且,Salifish是无比对的,比RSEM更节省时间。 RSEM也产生了良好的效果。
-
这估计是ERPKM没有RPKM流行的原因,因为effective transcript length 并没用什用,现在TPM准确性更高,使用的更频繁,TPM和RPKM计算公式相同。 -
TPM与RPKM的区别:唯一的不同是计算操作的顺序,TPM是先去除了基因长度的影响,而RPKM/FPKM是先去除测序深度的影响,TPM实际上改进了RPKM方法在不同样品间定量的不准确性。
-
-
-
然而,对于reads长度为76bp的样本,没有一种归一化方法改善相关结果。 因此,我们得出结论,当比对精度较高时,RC足以用于实际实验中的基因表达计算。 另外,Poly-A尾的影响实验是通过在转录本数据中添加腺嘌呤(0-25),结果表明,选择适当的poly-A尾部长度可以改善差异分析,但在本研究中似乎没有影响。 因此,我们建议研究者在基因差异表达分析的归一化步骤中不需要考虑ploy-a tails。
-
现在有参考基因组的物种比对精度都比较高,因为RNA-seq测序质量已经不是问题,因此可以使用 Row count 作为基因表达的计算,然后计算一下Fold Change,再取对数,比如我现在的这个课题就完全适用。 计算方法如下: E = mean(group1) B=mean(group2) FC = (E-B) / min(E,B)
-
参考文献
-
Li P, Piao Y, Shon HS, Ryu KH. Comparing the normalization methods for the differential analysis of Illumina high-throughput RNA-Seq data. BMC Bioinformatics. 2015 Oct 28;16:347. doi: 10.1186/s12859-015-0778-7. PMID: 26511205; PMCID: PMC4625728. -
deweylab/RSEM: RSEM: accurate quantification of gene and isoform expression from RNA-Seq data (github.com) -
【生信】【转录组】RSEM转录本定量 - 知乎 (zhihu.com)
本文由 mdnice 多平台发布

