
热门标签
热门文章
- 1Spark项目实训(一)_spark实训代码
- 2【Tokenizer原理篇】超详细!AIGC面试系列 大模型进阶(5)_大模型的tokenizer的实现方法及原理?
- 32D变换
- 4网络安全人士必备的30个安全工具_在网络安全方面,有哪些必备的安全软件和工具_网络安全中要掌握哪些常用的工具
- 5Agent Workflows(智能体工作流)
- 6EVE部署_部署eve
- 7Spark 各种配置项_spark.sql.storeassignmentpolicy
- 8【GitHub项目推荐--最佳开源TTS引擎】【转载】_github tts
- 9微信小程序获取微信用户绑定手机号--nodejs版本(测试账号,非非个人账号)_nodejs获取手机号
- 10linux 通过Java -jar 命令后台启动和关闭Java 程序_linux java -jar
当前位置: article > 正文
论文笔记:Highly accurate protein structure prediction with AlphaFold (AlphaFold 2 & appendix)
作者:码创造者 | 2024-06-25 22:34:08
赞
踩
highly accurate protein structure prediction with alphafold
注:这篇Nature的论文,如果光看正文,没法理解,需要结合它的补充材料一起看
同时这篇文章看得太吃力了QAQ,如果有说的不对的地方欢迎指正
0 前言
- 蛋白质结构预测:给定某一个蛋白质的一串氨基酸序列,猜测这个蛋白质的3D结构是什么样子的
- 现在的生物学可能需要很长的时间来具体了解一个蛋白质的结构
- 让蛋白质动起来,从不同的角度用显微镜来看它的结构
- 这篇论文提出了AlphaFold 2
- 前作AlphaFold 1精度不够
- AlphaFold 2的精度可以到达原子级别
- 实验室测得的(真实)位置和预测的位置之间的差距在原子大小的级别以内
- 模型使用了一定的生物学和物理学的知识,融合在深度学习里面
1 模型部分
1.1 整体模型

Transformer部分(也就是这里的encoder)只起到不同元素(氨基酸)之间信息整合的作用,真正信息的提炼部分,是在decoder部分实现的
我对于回收机制的想法:
这个机制有一点类似RNN中把hidden state再传给下一轮的RNN。通过不断地利用上一轮学到的输出,来获得更好的输出结果(每一轮的输出可能精度有限,通过不断迭代获得更好的效果)
区别在于,这里只重用结构,但是不回传梯度。(就是说传回去的几个输出是detach了的输出)
——>和RNN重用结构相比,虽然计算时间上没有区别,但是内存上是有区别的(RNN的话,“回传”的hidden state的梯度也是需要记录在内存中的,但是这边回传的输出是不用记录梯度的)
1.2 “encoder”部分
1.2.1 整体模型

1.2.2 row-wise gated self-attention with pair bias
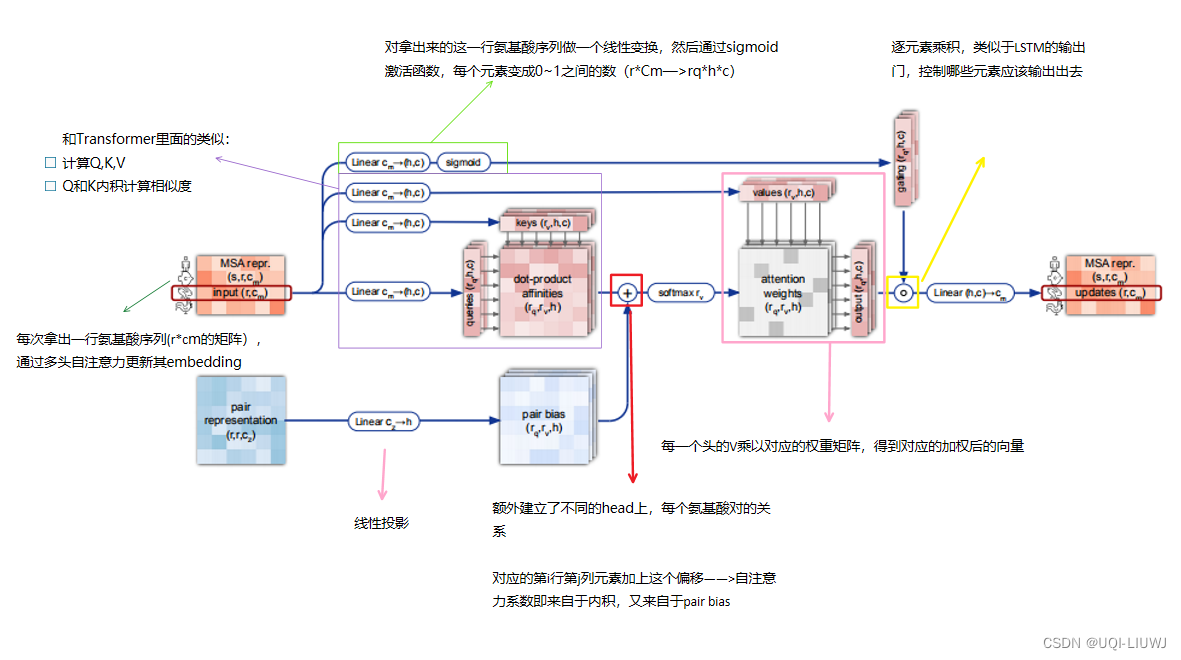

1.2.3 column-wise gated self-attention
大体流程和1.2.2 类似,区别在于,这里是按列来进行self-attention(同一个位置的氨基酸在不同蛋白质之中的权重)


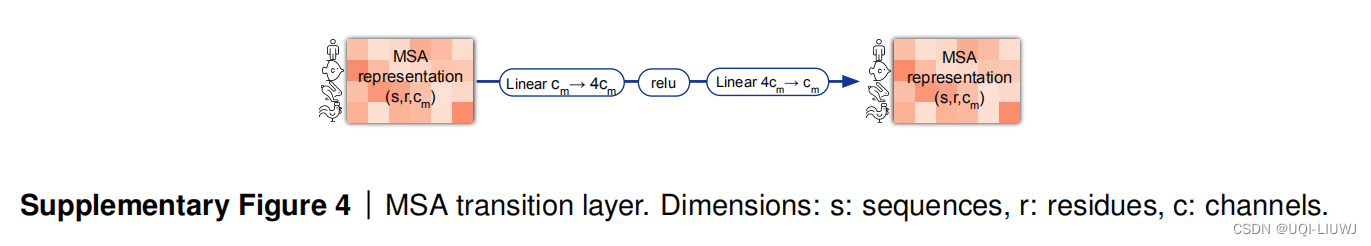
1.2.4 MSA transition
(两个transition是一样的)
这个就是一个MLP

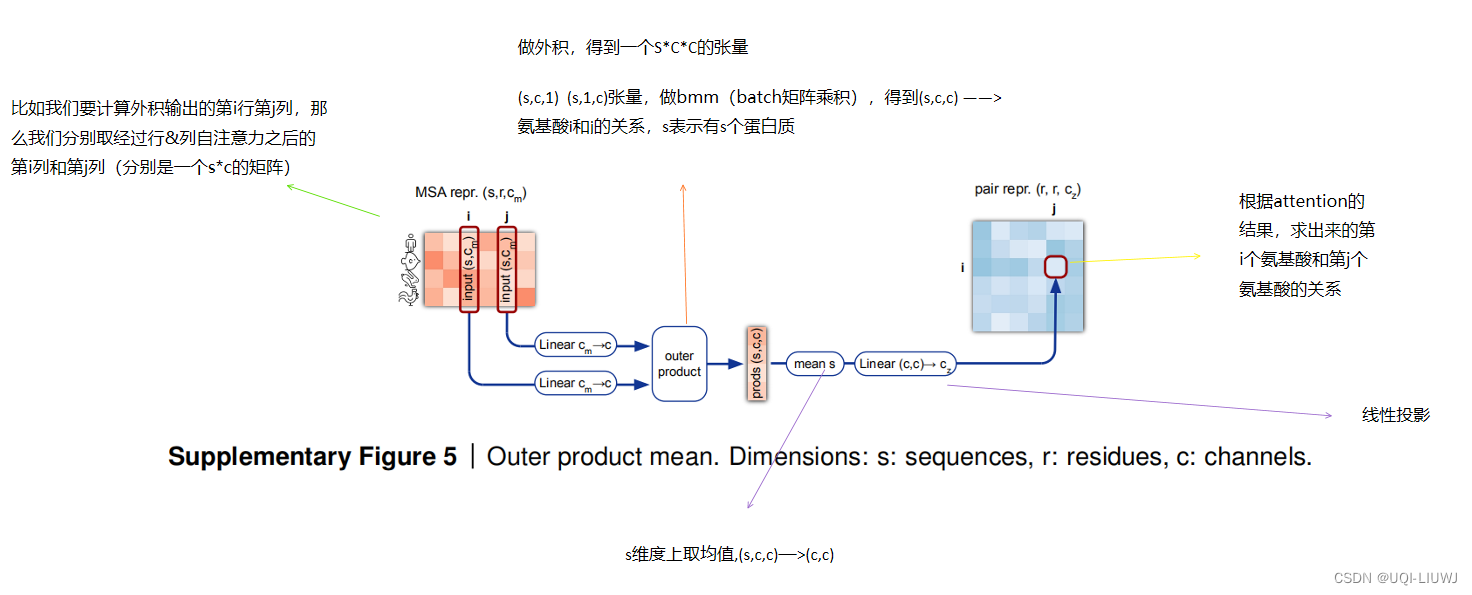
1.2.5 Outer product mean

1.2.6 Triangular multiplicative update
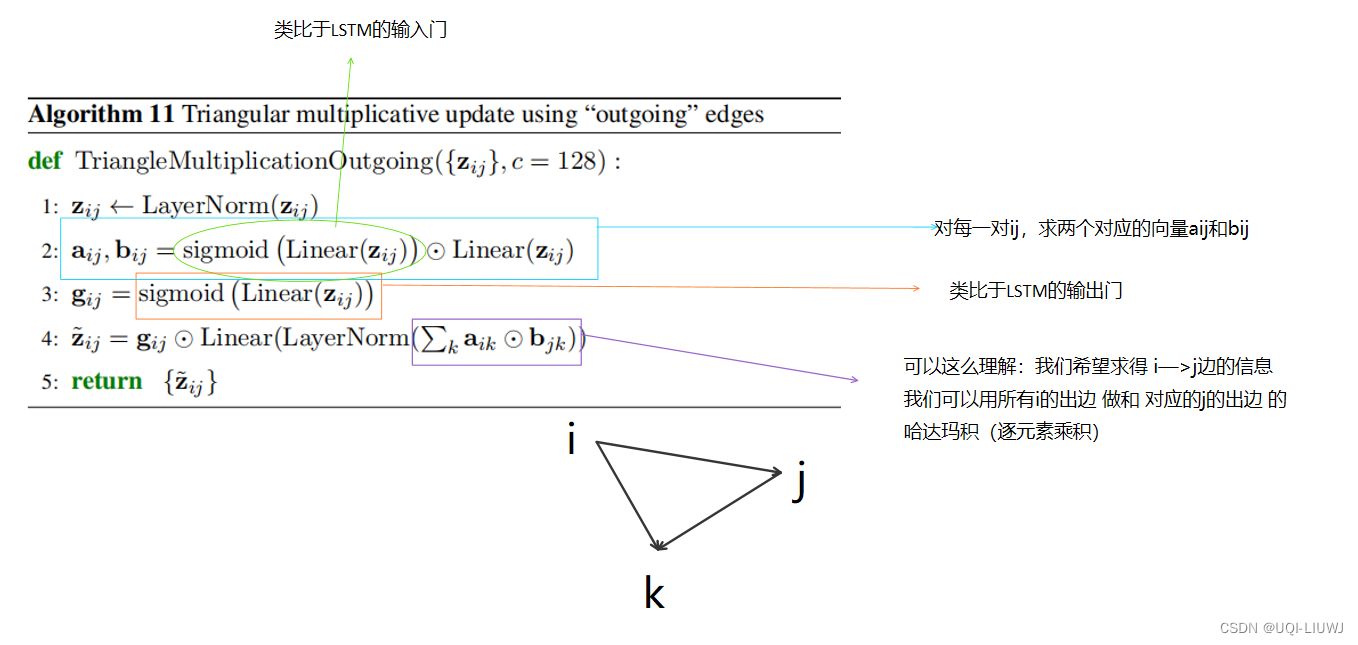
类似的,只是变成出边

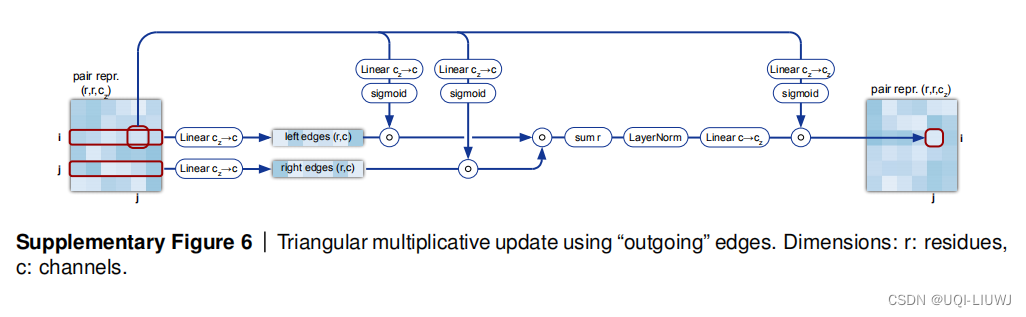
注意:由于出边入边模块的先后问题,所以得到的矩阵不一定对称。
1.2.7 Triangular self-attention
上图是之前的按行attention,下图是这里的attention,可以看到是很类似的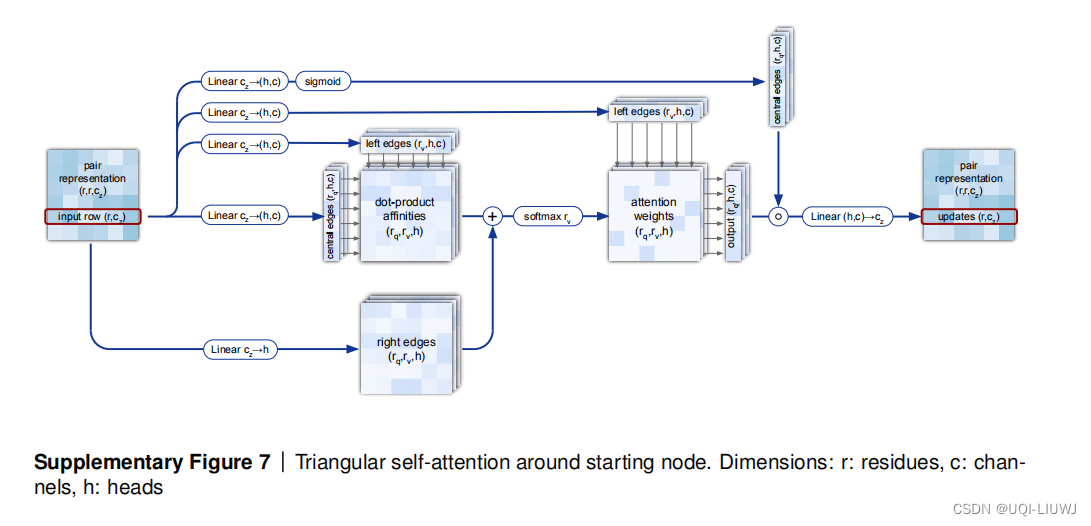

伪代码部分也基本一样,不过这里论文的意思是第五行(attention加权的部分)使用了一定的三角性质

1.3 “decoder”部分
1.3.0 如何预测?
- 表达蛋白质3D结构最简单的方法是记录每个元素的3D坐标。
- 蛋白质进行旋转/平移是不影响蛋白质结构的,但是如果用3D坐标的话,绝对位置会发生变化
- ——>所以这里使用的是相对位置
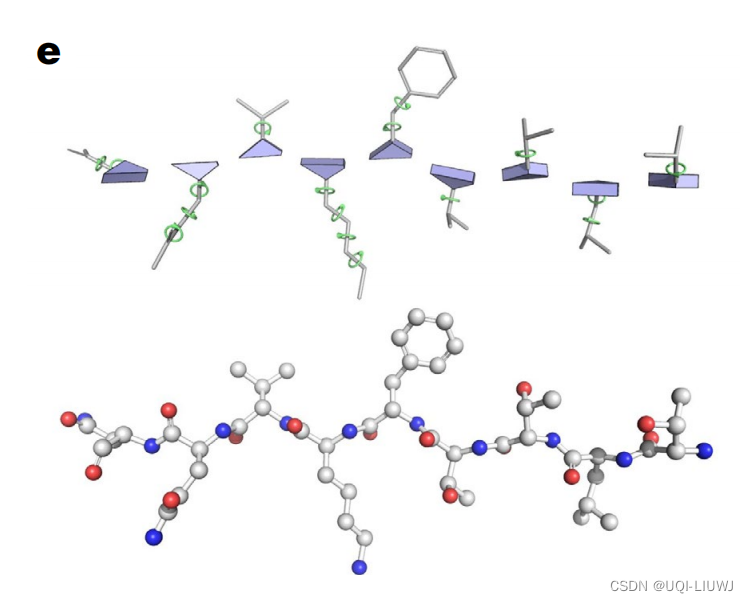
- 蛋白质可以想成主干+支链
- 主干点我们记为x,那么链上面任意一个点/主干的后一个点可以看成y=Rt+x
- 根据3*3的矩阵R做旋转
- 根据x做平移
1.3.1 整体模型
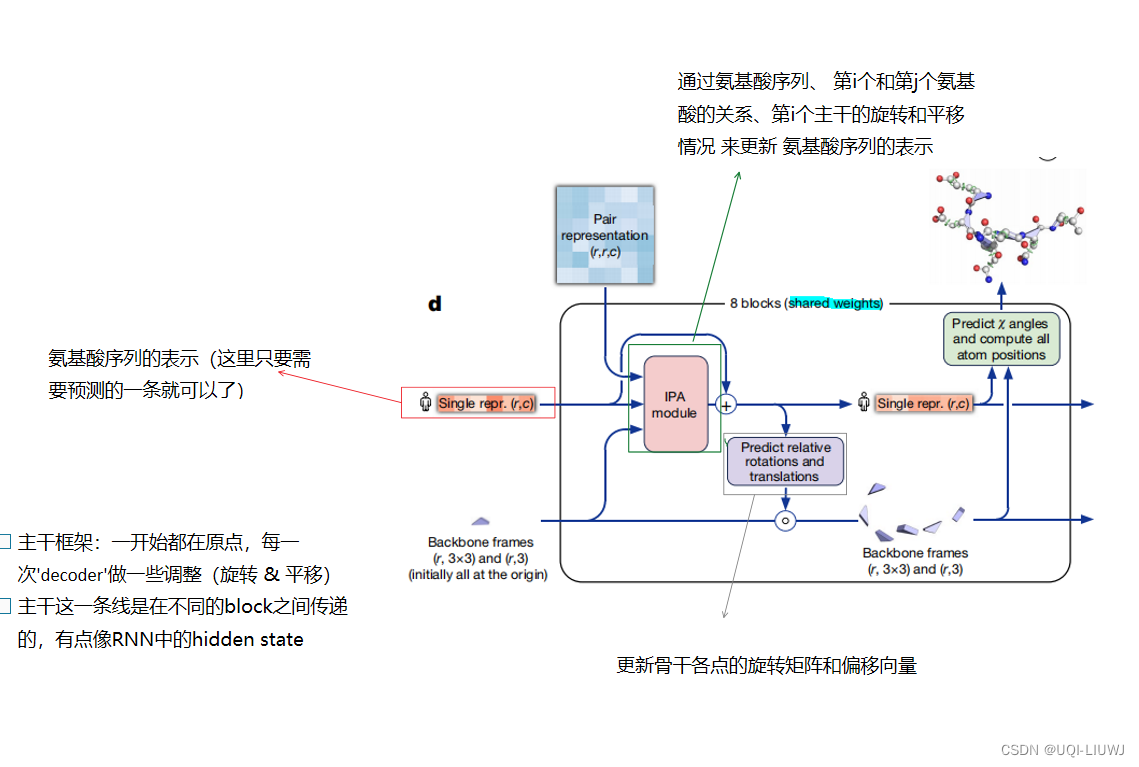
1.3.2 IPA Invariant point attention

1.3.3 Backbone update
更新骨干各点的s和T

实验效果
.1 和其他项目的对比

- 每一列是一个模型(参赛队伍)
- 每一条柱状图是对应的模型,平均预测位置和真实位置的区别(单位是
,即
米,也即原子的大小)
- 可以看到AlphaFold 2的精度已经达到了原子精度,这是一个里程碑意义的精度
2 AlphaFold 预测的精度

- 蓝色的是AlphaFold 预测的结果
- 绿色的是实验室预测出来的结果
- 可以看到他们的误差确实在一个原子的大小(黑色球体)以内
声明:本文内容由网友自发贡献,转载请注明出处:【wpsshop】
推荐阅读
相关标签

