- 1Xeno RAT成为GitHub上的严重威胁;黑客组织Lazarus利用Windows内核0day漏洞;五眼联盟警告 Ivanti漏洞被广泛利用 | 安全周报 0301
- 2一键ai绘画软件哪个好?分享几款一键ai绘画手机版给你_一键画画
- 3CLUE--汉语语言理解评估基准_clue基准
- 4C++ 中的 std::vector:动态数组操作和常用函数解析
- 5深入解析CSS单位:全面理解与实战应用
- 6Kaggle入门比赛:灾难推文的自然语言处理 详细教程_kaggle自然语言处理入门项目
- 72020年计算机设计大赛参赛回顾与总结_计算机设计大赛提交作品需要准备些什么
- 8javax.net.ssl.SSLHandshakeException: sun.security.validator.ValidatorException: PKIX path building f
- 9基于Java(SpringBoot框架)毕业设计作品成品(38)高校校园疫情防控系统设计与实现
- 10【C++】蓝桥杯第十二届省赛杨辉三角形(小白易懂)_蓝桥杯杨辉三角形
windows ubuntu 子系统:肿瘤全外篇,bam质控
赞
踩
各个环节的质控,
raw和clean都要质控,
比对的各环节的bam文件都要质控,
使用qulima对wes的比对bam文件总结测序深度及覆盖率。
samtools flagstat L1_recalibrated_reads.bam
该命令将输出 BAM 文件的一些统计信息,包括总读取数、比对上参考序列的读取数、比对到不同位置的读取数等。
#结果可如下。
L1_recalibrated_reads.bam 的统计信息如下:
总读取数:103,094,432
比对上参考序列的读取数:103,028,917 (占总读取数的 99.94%)
次要比对的 reads 数:0
补充比对的 reads 数:674,520
重复 reads 数:22,411,852
成对测序的 reads 数:102,419,912
测序的 read1 数:51,209,956
测序的 read2 数:51,209,956
正确成对匹配的 reads 数:101,697,064 (占成对测序的 reads 的 99.29%)
自身及其 mate 均比对到参考序列的 reads 数:102,306,392
单独出现的 reads 数:48,005 (占总读取数的 0.05%)
与不同染色体的 mate 均比对的 reads 数:400,816
映射到不同染色体且 mapQ 值大于等于 5 的 reads 数:304,376
运行以下命令可以计算 L1.bam 中的总行数(即记录数),从而得知该 BAM 文件中包含多少条比对信息:samtools view 949743-T_L2_1.bam | wc -l
#获取全外bed文件
CCDS官网
进入官网后进入其ftp服务器
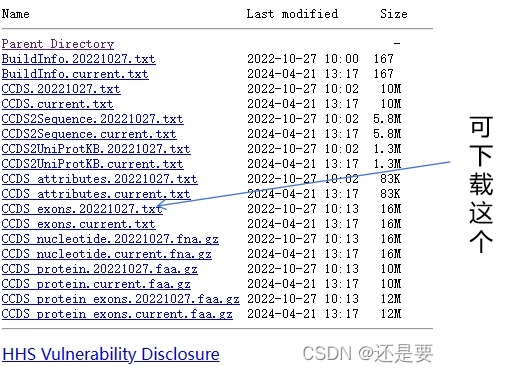
cat CCDS.20221027.txt | perl -alne '{/
这条命令的作用是将 CCDS(Consensus CDS)数据中的 exons 信息提取出来,生成一个 BED 文件 hg38.exon.bed。具体实现步骤如下:使用 cat 命令将 CCDS.20221027.txt 文件的内容输出到标准输出。
使用 perl 命令解析每一行,并通过正则表达式提取出 exons 信息。如果该行不包含 exons 信息,则跳过。
将提取到的 exons 信息进行格式化,并使用 split 函数将其拆分成多个 exon。对于每个 exon,输出其所在的染色体、起始位置、终止位置和所属基因。
使用 sort 命令将输出结果按照染色体、起始位置和终止位置排序。
使用 awk 命令将排序后的结果转换为 BED 格式,并指定其 score 和 strand 信息,最终将结果输出到 hg38.exon.bed 文件中。
这个 hg38.exon.bed 文件可以用于基因组注释和区域相关的分析。
samtools view L1_recalibrated_reads.bam | less -S
这条命令使用 samtools view 命令来查看 949743-T_L2_1_recalibrated_reads.bam 这个 BAM 文件的内容,并通过管道将输出传递给 less -S 命令进行分页查看。
samtools view 命令用于从 BAM 文件中读取比对信息,并以文本格式输出。| 符号表示将前一个命令的输出作为后一个命令的输入进行处理。
less 命令是一个分页查看器,可以按需滚动查看文件的内容。-S 参数用于禁用行内过长时的折行显示,保持每行内容在屏幕上的可见性。
因此,执行该命令后,将能够使用 less 分页查看 L1_recalibrated_reads.bam 文件中的比对信息。您可以使用方向键(上下左右)和 Page Up/Page Down 键来浏览文件内容,并使用 q 键退出 less 查看器。
# 1. 创建输出目录
mkdir -p qc_results
#安装qualimap
qualimap bamqc \
-bam L1.bam \
-outdir qc_results \
-c \
--java-mem-size=4G \
--feature-file /mnt/h/db/hg38.bed/hg38.exon.bed \
-nt 4
qualimap bamqc: 这是运行 Qualimap 工具中的 bamqc 模块的命令,用于评估 BAM 文件的质量。
-bam L1.bam: -bam 参数指定输入的 BAM 文件,这里使用的是 949743-T_L2_1.bam 文件。
-outdir qc_results: -outdir 参数指定输出结果的目录,这里结果将保存在名为 qc_results 的目录中。
-c: -c 参数表示生成覆盖度报告。
--java-mem-size=4G: --java-mem-size 参数指定分配给 Java 虚拟机的内存大小,这里设置为 4GB。
--feature-file /mnt/h/db/hg38.bed/hg38.exon.bed: --feature-file 参数指定感兴趣的区域文件,这里使用的是一个 BED 格式的文件,其中包含了人类基因组 hg38 版本的外显子区域信息。
-nt 4: -nt 参数指定并行运行的线程数,这里设置为 4 个线程。
出来以下结果,有些难懂。
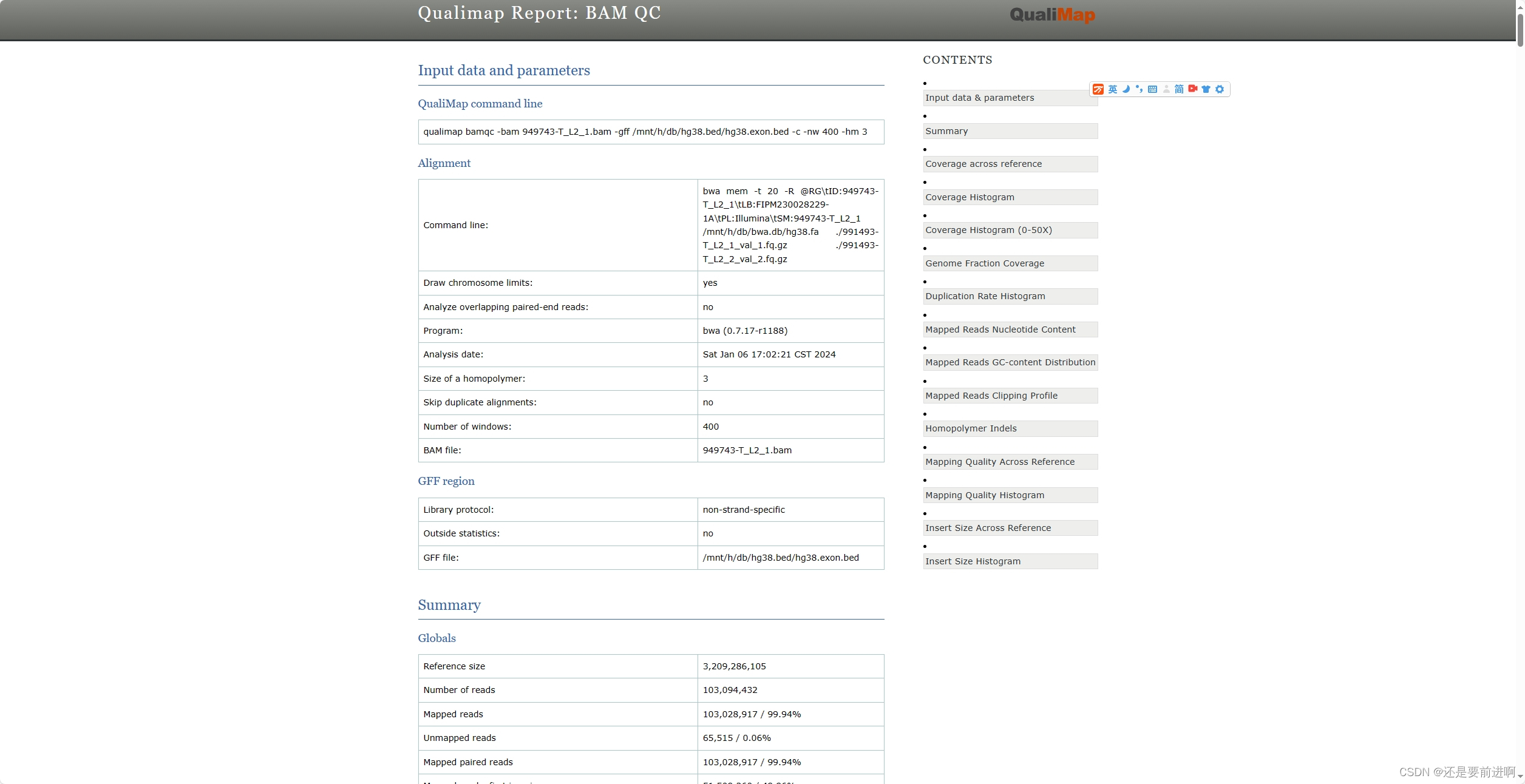
可用multiqc整理一下就好看多了。