
- 1Eureka服务注册源码分析_initialstatus: down
- 2BP神经网络python实现_bp神经网络多变量时间预测实例 python
- 32024年保护微服务的前10种技术
- 4语音分离论文:Conv-TasNet:Surpassing Ideal Time–Frequency Magnitude Masking for Speech Separation_时域语音分离论文
- 5多模态模型技术综述_多模态数据算法公式
- 6【对抗攻击代码实战】对抗样本的生成——FGSM_对抗样本生成
- 7SpringBoot SpringBoot 基础篇 2 SpringBoot 基础配置 2.8 yaml 文件中的变量引用_yml文件引用变量java
- 8vivado2019.1关联modelsim仿真_vivado modelsim联合仿真
- 9Pytorch搭建全卷积网络训练mstar数据集
- 10最完整的全球生成式AI生态地图;900+LLM开源工具清单与我的观察;我开发了Devin平替,6个月的血泪经验;月之暗面新一轮内测 | ShowMeAI日报
ProtTrans-Glutar: 整合来自Pre-trained Transformer-Based Models的特征用于预测谷氨酰化位点【Frontiers in Genetics,2022】_多肽库预测酰化位点
赞
踩
研究背景:
- 氨基酸的 PTM (post-translational modification) 能够改变蛋白的功能,其中赖氨酸谷氨酰化修饰 (lysine glutarylation) 指的是谷氨酰基团连接到赖氨酸残基上,这种修饰可以通过免疫印迹和质谱分析等生化方法进行鉴定。
- 赖氨酸谷氨酰化修饰可能是衰老和细胞压力的一种分子标记,谷氨酰化修饰的紊乱与一些代谢疾病(比如 I型戊二酸血症、糖尿病、癌症等)相关。
- 用蛋白组技术鉴定谷氨酰化修饰的多肽是昂贵且耗时的,所以用计算方法来快速鉴定谷氨酰化是很重要的。
- 先前研究已经提出了多种预测谷氨酰化位点的模型,比如 GluPred、iGlu-Lys、MDDGlutar、RF-GlutarySite、Iglu_Adaboost等,尽管这些模型都被用于鉴定谷氨酰化位点,但是预测效果有限,主要问题是这些模型不能有效获取蛋白质序列的特征。
- BERT或其他的基于Transformer的语言模型在NLP领域取得了巨大成功,而且之前研究表明用蛋白序列对BERT或者BERT-like模型进行预训练,能够有效地捕获序列特征用于蛋白分类,所以用 pre-trained BERT 对蛋白序列进行 embedding 也许能够构建更好的谷氨酰化修饰预测模型。
- 本论文中,作者提出了一个新的预测谷氨酰化修饰位点的模型,它利用预训练的蛋白模型获取序列特征,并与传统方法获取的序列特征对比,表明基于 pre-trained model 的方法能够更有效的获取序列特征,提升预测效果。
研究目的:
利用多肽序列特征来预测多肽片段中的中心赖氨酸残基是否会被谷氨酰化修饰。
数据集构成:
unbalanced benchmark dataset compiled by Al-barakati et al., 2019
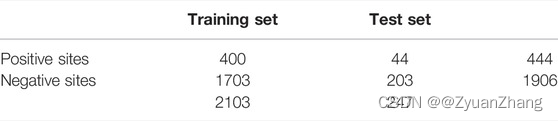
- 数据集包含多个来源总共234个蛋白的749个位点,包括 PLMD (Xu et al., 2017) 和 (Tan et al., 2014),涉及4个不同的物种(Mus musculus, Mycobacterium tuberculosis, E. coli, and HeLa cells)
- 将数据集中的蛋白序列用CD-HIT取冗余(相似性阈值为0.4),并将剩下的蛋白切成长度为23的多肽,同时确保谷氨酰化赖氨酸位于中间位置。负样本做同样处理,但是中心赖氨酸残基未被谷氨酰化修饰。
- 经上述处理之后,数据集构成如下所示:
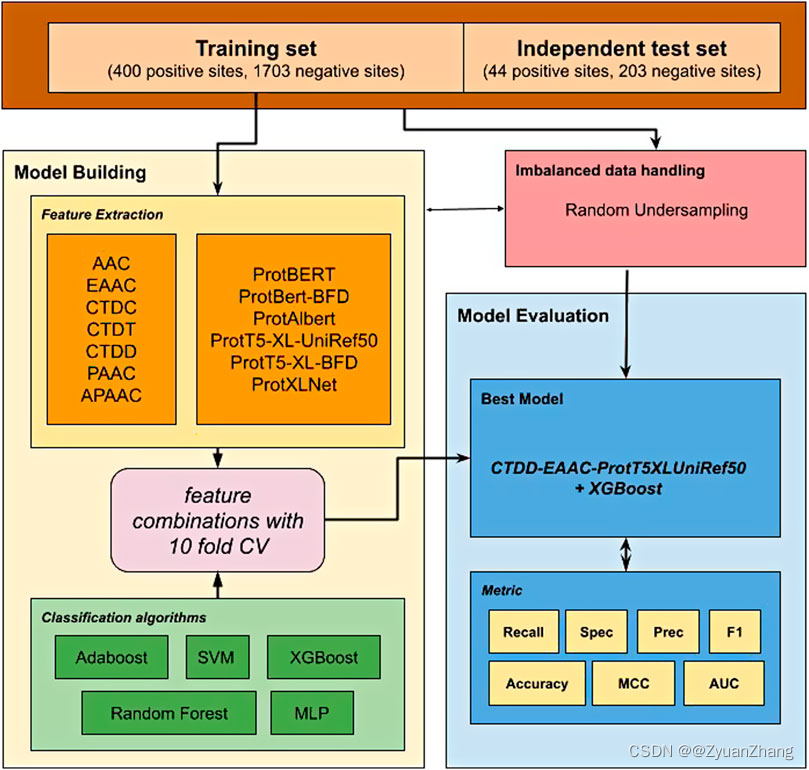
研究思路和方法:
论文代码:https://github.com/findriani/ProtTrans-Glutar
1. Feature Extraction:
Feature Extraction 包含 classic sequence-based protein features 和 features derived from pre-trained transformer-based protein embedding
输入文件中蛋白序列的格式(以作者提供的 sample.fasta为例,这些序列的中心氨基酸多是赖氨酸):
>postest1 SLVNELTFSARKMMADEALDSGL >postest2 PSVSLQTVKLMKEGLEAARLKAY >postest3 LMDCMNKLKNNKEYLEFRKERSK >postest4 AATILTSPDLRKQWLQEVKGMAD >postest5 GLKPEQVERLTKEFSVYMTKDGR >negtest1 GGVEFNIDLPNKKVCIDSEHSSD >negtest2 EQNRTHSASFFKFLTEELSLDQD >negtest3 AFWRELVECFQKISKDSDCRAVV >negtest4 KIFRQQLEVFMKKNVDFLIAEYF >negtest5 GSWGSGLDMHTKPWIRARARKEY

- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
1.1 classic sequence-based protein features:
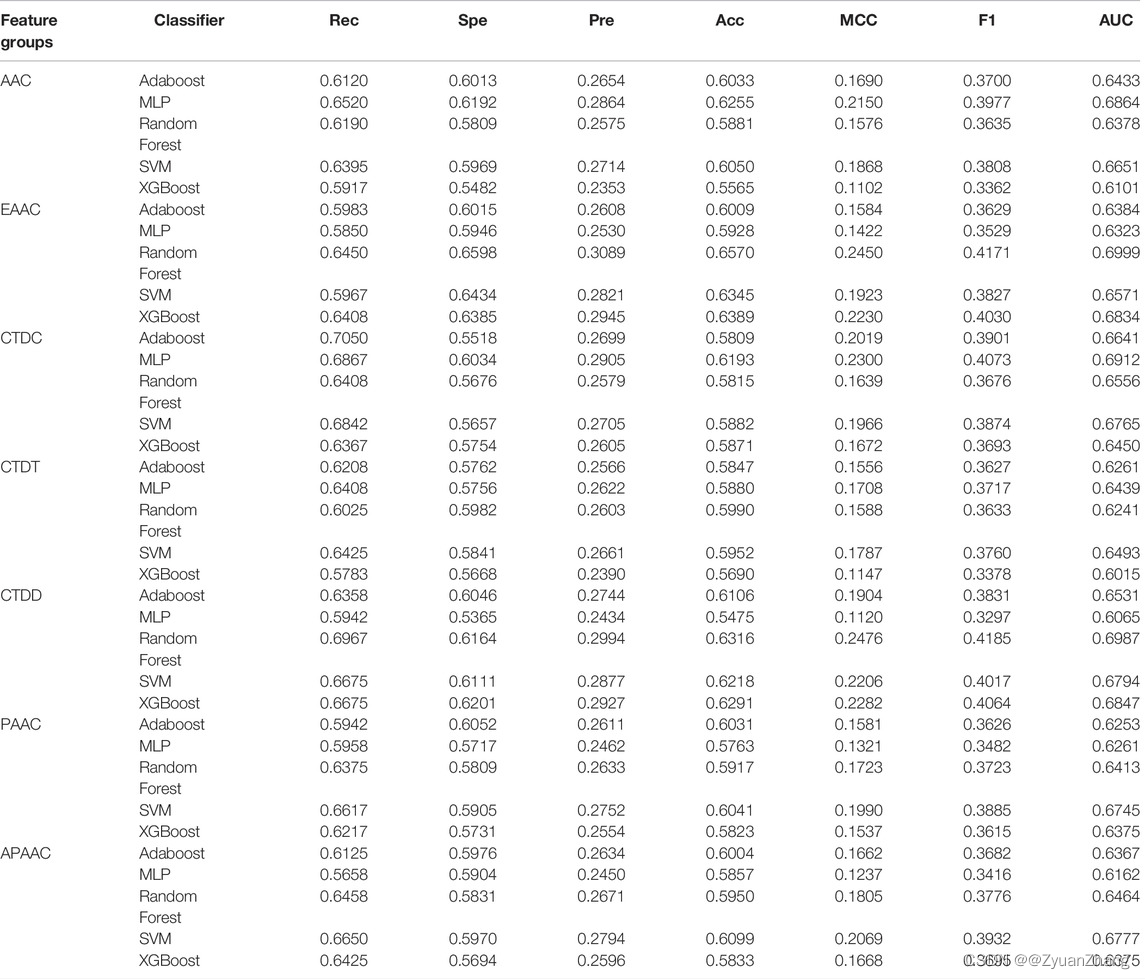
经典的序列特征由 iFeature 获取,这些特征包括 AAC、EAAC、Composition、Transition、Distribution、PAAC 和 APAAC。
这部分直接运行
iFeature.py即可,通过参数--type来指定需要获取哪种序列特征,如下所示:


关于AAC、EAAD等的简要解释(详情请见相关论文):
AAC: Amino Acid Composition,计算一条序列中每种氨基酸出现的频率,得到一个长度为20的向量。
EAAD: Enhanced Amino Acid Composition,计算一个移动窗口内,氨基酸出现的频率。
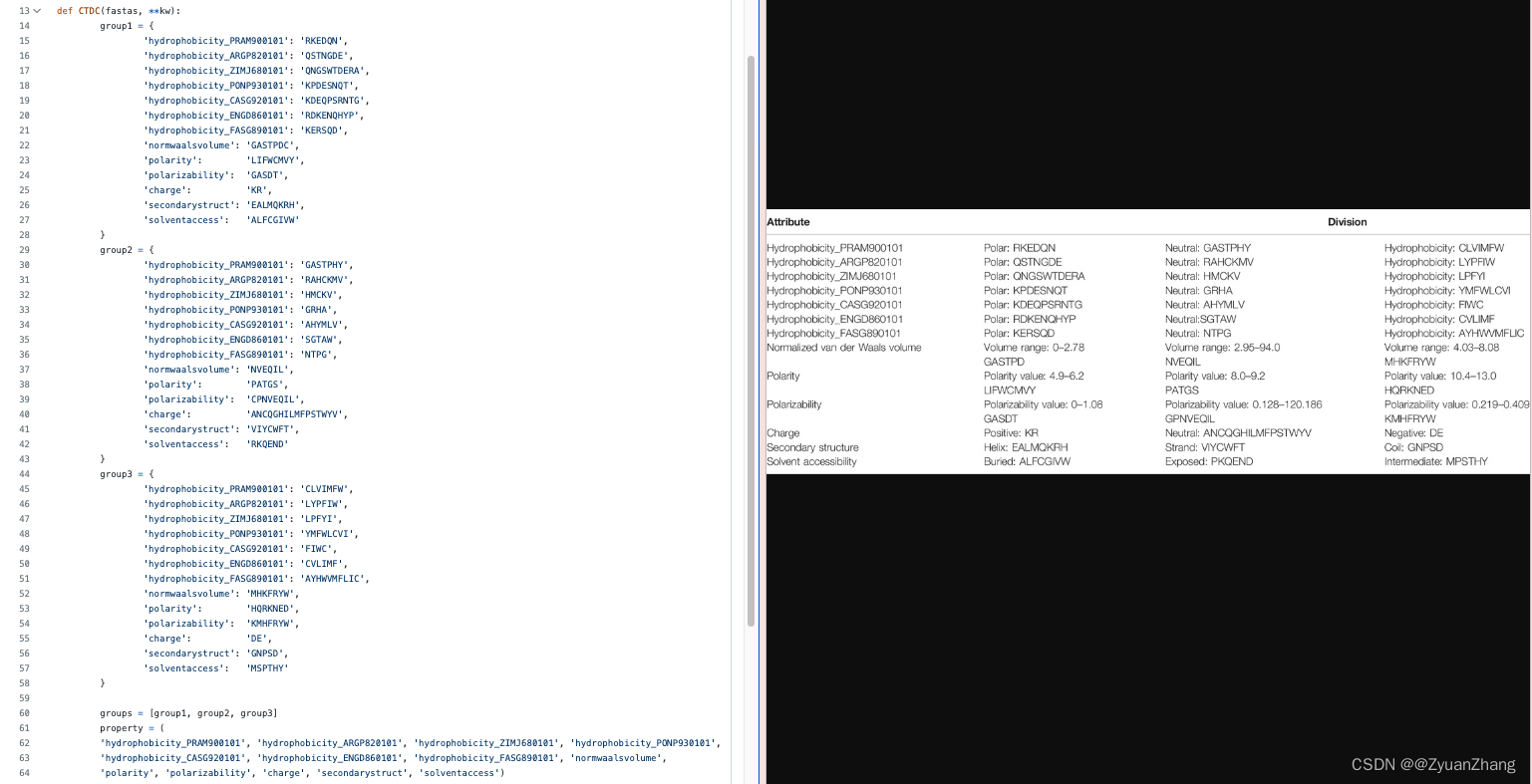
根据氨基酸的理化性质,将氨基酸分为三组(如下图所示):
Composition: 将氨基酸序列用三个group替换之后,计算每个group出现的频率。
Transition: 将氨基酸序列用三个group替换之后,计算 “rs” (二肽) 出现的频率。
Distribution: 将氨基酸序列用三个group替换之后,计算 first occurrence, 25%, 50%, 75%, 和 100%序列长度中每个group出现的频率。
与AAC类似,PAAC也是使用氨基酸频率矩阵来表征蛋白,但与AAC不同的是PAAC增加了一些额外的信息(如 一定距离的残基之间的相关性)。APAAC是PAAC的一种变体。
1.2 features derived from pre-trained transformer-based protein embedding:
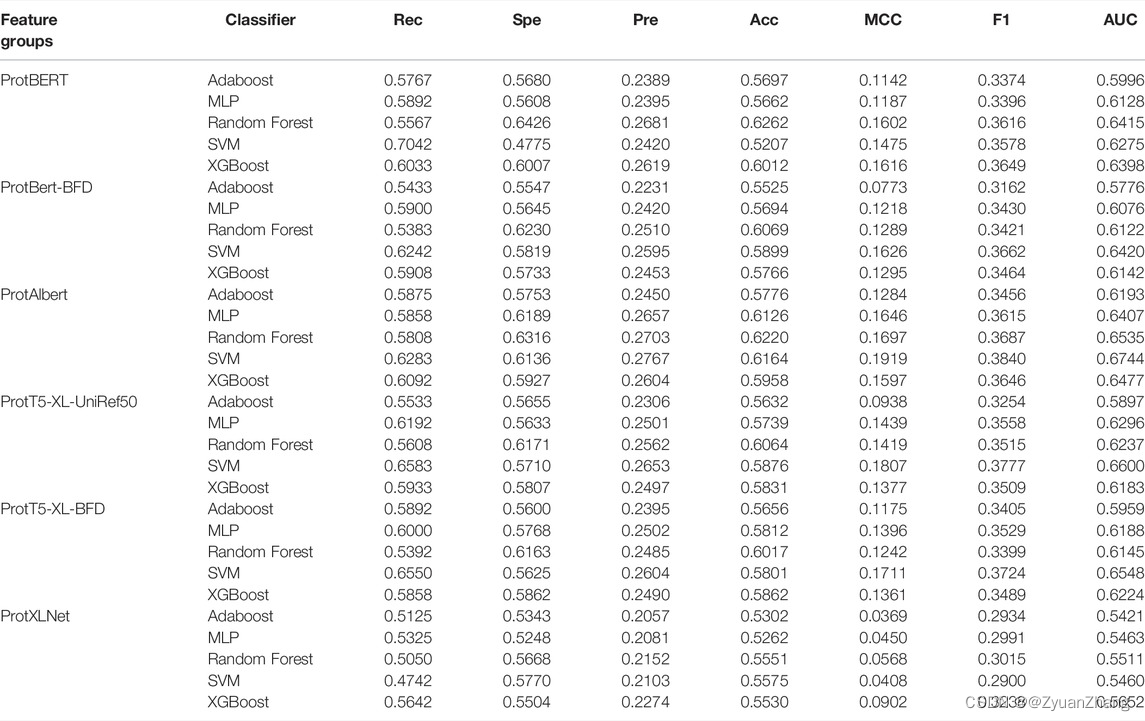
用到的预训练 transformer 模型有6个:ProtBERT、ProtBert-BFD、ProtAlbert、ProtT5-XL-UniRef50、ProtT5-XL-BFD 和 ProtXLNet。
这些预训练模型可以从 https://huggingface.co/Rostlab 上下载(如下图所示):
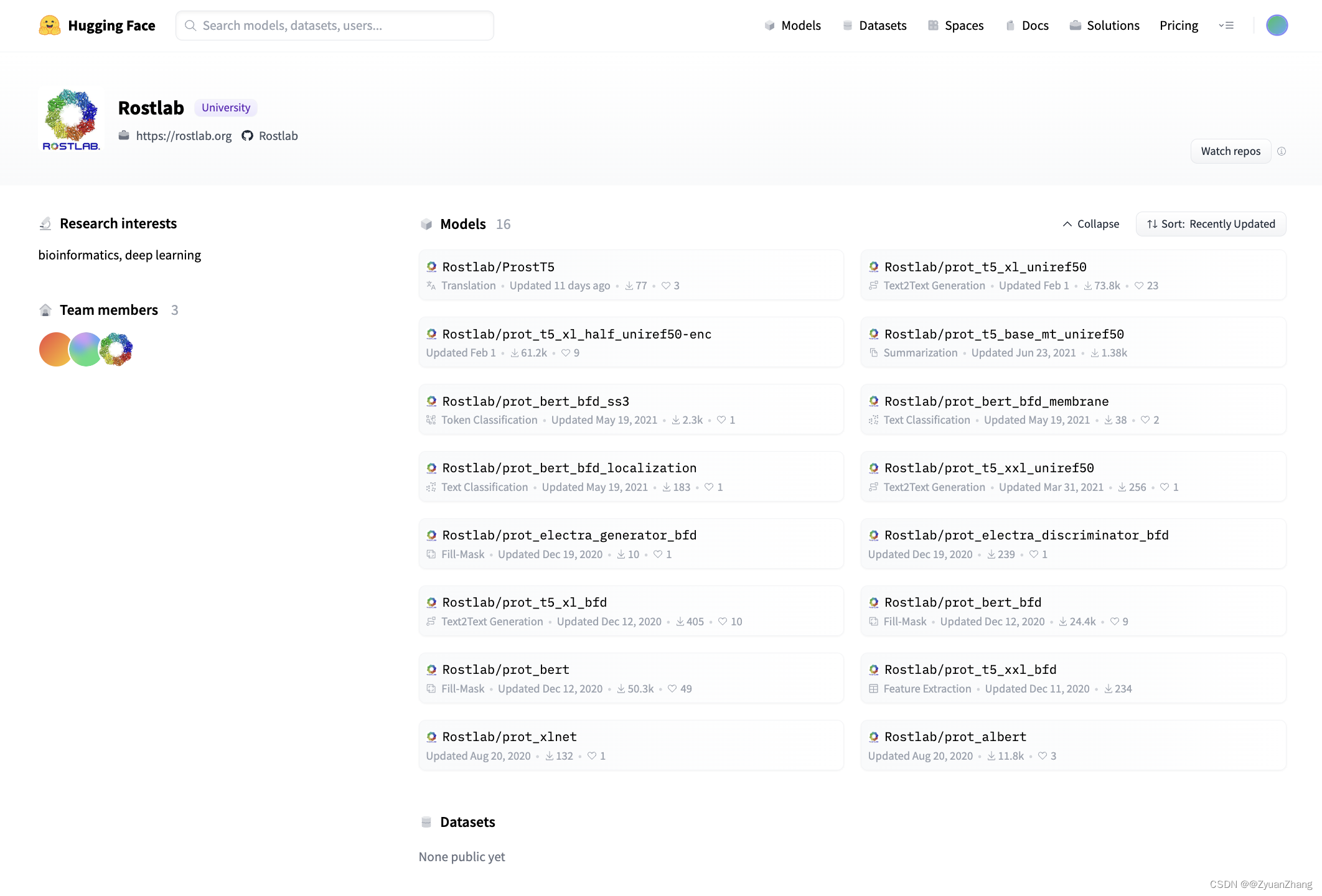
1.2.1 加载预训练 Transformer 模型:
通过修改Rostlab/prot_t5_xl_uniref50名称来指定对应的预训练 Transformer 模型。
关于如何下载 huggingface-transformers 模型,可以参考:https://zhuanlan.zhihu.com/p/475260268
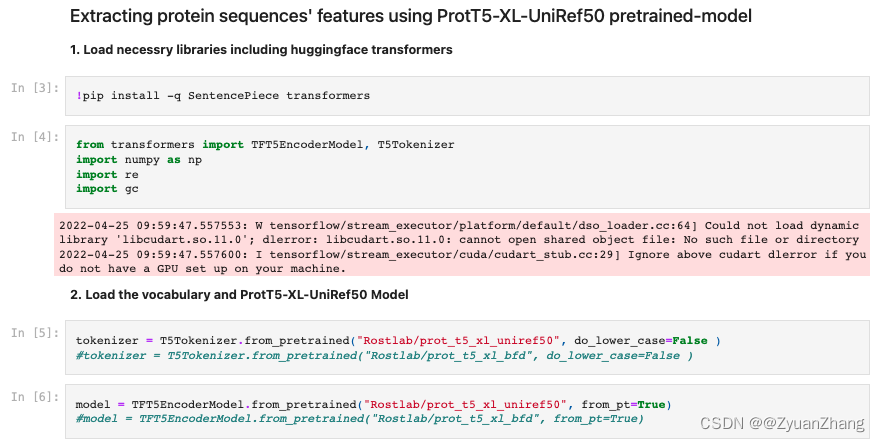
1.2.2 加载蛋白序列:
读取 sample.fasta 文件,按照字符对序列进行分词,并将这些蛋白序列中(U, Z, O, B)换成 X(如下图所示)。
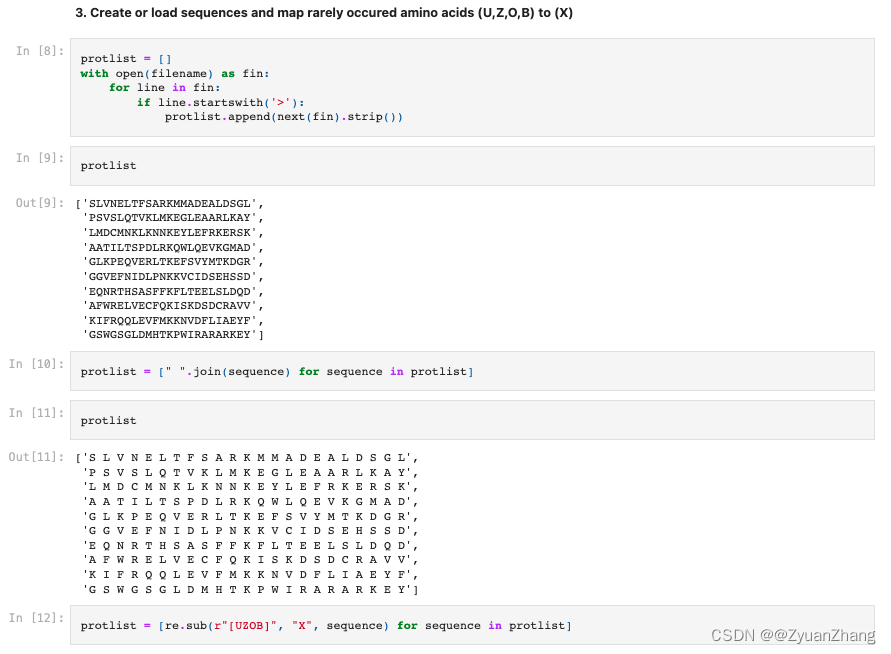
1.2.3 对蛋白序列进行编码:
将字符转化为 tokenizer 中的索引。
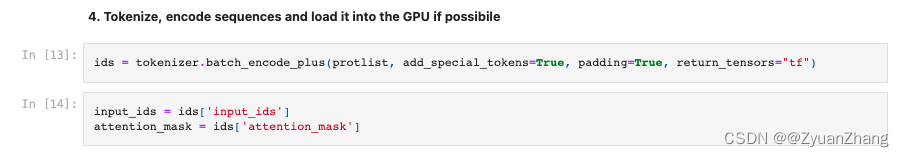
1.2.4 获取序列的特征:

1.2.5 去掉填充部分和特殊字符的特征行:
因为对序列进行编码的时候
add_special_tokens=True, padding=True,所以这一步需要去掉填充和特殊字符。
每条序列都被编码为形状为 nx1024 的特征矩阵(n为序列长度,sample.fasta中的多肽序列长度为23,对应的特征矩阵形状为 23x1024),可以通过features[0].shape查看第一条序列的特征形状。

1.2.6 将每条序列对应的特征矩阵按照特征列进行加和:
假设features.shape = (100, 23, 1024),分别对应(序列数目, 每条序列的长度, 每个位置的氨基酸对应的特征向量),经过这一步处理之后,prott5_xl_uniref50.shape = (100, 1024)。
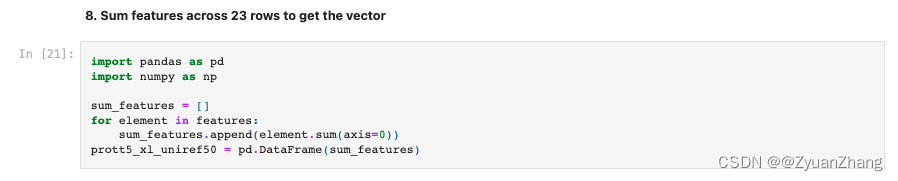
2. Feature Combinations:
其中ctdd、eaac和prott5_xl_uniref50的shape[0]是一样的,都是蛋白序列的数目。

3. Classification modeling and select the best one:
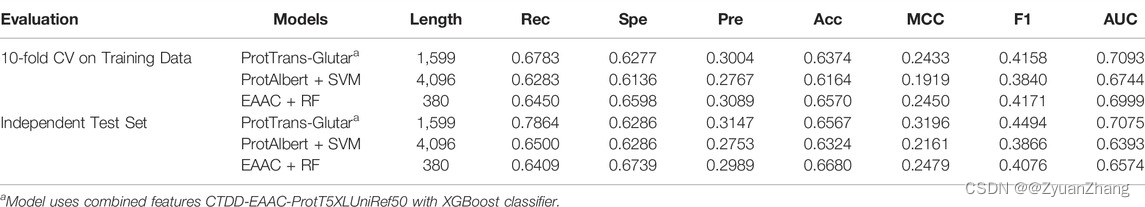
所用的机器学习模型包括 Adaboost、SVM、XGBoost、Random Forest 和 MLP,分别进行训练建模,并进行 10-fold-cv,从中选取最优的模型(CTDD-EACC-ProtT5XLUniRef50 + XGBoost),并计算对应的 Accuracy、Precision 等指标。
实验结果及讨论:
1. 基于序列特征的交叉验证结果:
2. 基于 pre-trained transformer models 的交叉验证结果:
3. 基于特征整合的最优的模型结果:
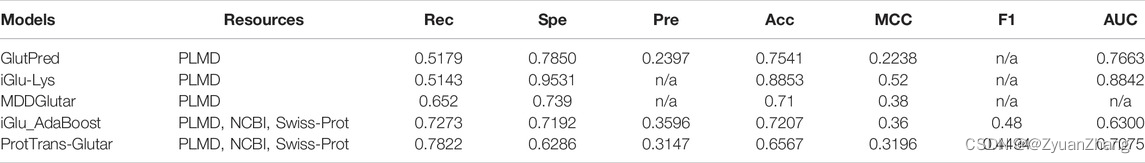
4. 和现有方法预测效果的比较:
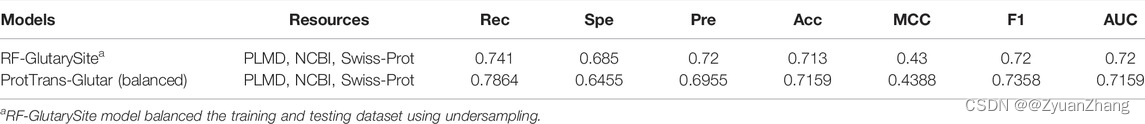
5. 在类别平衡的数据集上模型的性能:
对于 imbalanced data 的处理方法:random under-sampling strategy。
6. 讨论:
- 无论基于传统方法得到的序列特征还是预训练模型得到的序列特征,它们各自单独用于训练模型,得到的模型效果都是比较差的,但是当把它们整合到一起时,可以得到更好的模型性能。
- 通过与现有的一些用于预测赖氨酸谷氨酰化修饰的方法相比,作者提出的 ProtTrans-Glutar 模型的效果是更好的,而如果使用类别均衡的数据集,该模型与之前的模型 RF-GlutarySite 的性能是差不多的。
- 整体上而言,受到谷氨酰化修饰的数据限制,传统序列特征提取方法和预训练模型提取特征的方法相结合,可以提升 Recall 和 AUC,但是 Specificity 并没有多少变化。
- 综上所述,无论是传统方法还是预训练模型,获取蛋白质序列特征的效果是有限的,未来仍需要一些新的更有效的方法。

