
- 1几种常见的Java单例模式写法_单例模式aobing
- 2安装时出现Requirement already satisfied问题_安装时报requierements satif
- 3c#label控件_c# label
- 4基于OpenMV和STM32F103C8T6的视觉巡线小车_使用openmv摄像头还需要stm32还需要
- 5YOLOv3(keras)训练自己的数据集(voc)_训练voc图片
- 6女生学测试还是前端好?_女生做测试好还是前端开发好
- 71每天Python小例-12306爬虫#WinError 2_爬虫winerror 2
- 8边缘计算(Edge Computing)_关键概念/优势/应用场景
- 9KOSMOS-2.5:密集文本的多模态读写模型_kosmos2.5
- 10国产算力芯片排行榜前十名_国产ai算力芯片公司排行
隐藏在发表的宏基因组文章背后故事,如何发掘和学习_宏基因组队列文章
赞
踩
近年来,随着测序技术的发展,对微生物群(微生物组)的研究逐渐加深,研究热点越来越多集中于环境和生物体相互作用的微生物群。加之测序成本降低,分析技术不断提升,都使得宏基因组测序技术得到广泛应用。
为什么要做宏基因组
宏基因组相对16S来说其物种分辨率会更高,随着物种测序完成越来越多,数据库更加完善,在肠道菌群方面基本能实现97%以上的菌都能鉴定到种,90%以上到菌株层面。
而且可以同时获得除RNA病毒外的所有物种的分布。此外包括菌基因组CNV等方法的出现,可以直接通过大规模宏基因组测序不仅找到可能的菌,进一步还能鉴定出特定候选基因区段。
对16s而言,宏基因组可挖掘的空间更大,如果想提高文章深度,发更高分的文章,宏基因组是个不错的选择。
有人说,宏基因组虽然有深度,但相应也更加复杂化。比如同样是做某类疾病,别人通过实验测序分析可以得出结论写出文章,到自己去研究怎么就遇到各种困难?完全没差异?出不了结果?
......
而等到所有分析完重新回头去找问题,也很难发现问题所在,一遍遍重新做耗时耗力...
其实这类文章并不只是测序而已,重点在于理解这个过程,研究方向的思考,分析策略的选择,包括其中要注意的因素等。
本文将从一个例子开始,为你打开研究思路,绕过某些易踩的坑,让你的数据更有利用价值,处理具体的问题也更加得心应手。
宏基因组文章解析
下面是利用已发表的宏基因组文章中的数据,用我们自己的流程进行一次全面的分析。在这个过程中,我们发现很多有意思的点,分享给大家。

这是一篇做精神分裂症患者粪便菌群宏基因组的文章。文中分析了90名未经药物治疗的精神分裂症患者和81名健康对照者的粪便菌群,确定了一种能够区分患者和对照者的微生物物种分类器。
研究发现与精神分裂症相关的功能代谢方面的差异主要体现在短链脂肪酸合成、色氨酸代谢以及神经递质的合成/降解。还发现了一种在精神分裂症中比较富集的物种 Streptococcus vestibularis,在小鼠的粪移植实验中,它引起了小鼠的社交行为缺陷,并改变了小鼠外周组织中神经递质的水平。
我们对这篇文章中的肠-脑模块(GBM)部分的分析比较感兴趣。为此,下载了文中使用的宏基因组原始数据,原计171个样本,但有一个无法下载成功(所以结果方面可能会与原作有细微的差异)。
元数据的收集
在宏基因组分析之前通常要先收集元数据,那么什么是元数据?
元 数 据
元数据主要是对数据的属性进行描述的数据,也是实验数据的重要组成部分。
人体样本:主要包括个人基本生理信息(例如身高、体重、年龄、性别等)、生活行为方式、地理位置、膳食结构、营养状况、既往病史、抗生素使用情况等信息。
环境样本:指样本获取过程中的信息,例如采集地点、大气、水文、温度、pH值、压力、季节、运输方法、存储媒介等。
在数据分析之前我们首先对这批样本的群体构成和一些已有的元数据进行统计和特征分析:

其中,SCZ是精神分裂症患者,HC是健康对照。
一共170例样本,其中精神病患者90例。
年龄、性别以及BMI还有食物构成的分布情况如下:

从上图中可以看出有些元信息在分组间就存在明显差异,如年龄,血清素指标,而有些元信息如性别没有明显差异。
进一步对这些元信息做统计分析,发现健康组(HC)和精神病患者(SCZ)存在显著差异(如下图的年龄信息,P值为0.117)。在我们的宏基因组分析流程中,分析前会将客户提供的所有样本元信息做统计分析,作为进一步分析的基础。

其中除了性别数据,其他几个主要因素还是存在差异的,这就要求后续分析的时候需要考虑这些因素的影响,这个在后面会提到。
研究中同时检测了血清中主要神经递质的水平,可以从中看出精神病患者的神经递质差异特征,其中多项神经递质存在极其明显的差异:

上图中框出的色氨酸、谷氨酸、酪氨酸、苯丙氨酸都存在显著差异。
数据库的选择和完善程度
我们使用的是kraken2以及谷禾自建的基于Reseq 99版本的微生物基因组数据库。需要注意的是Kraken2自带的物种数据库,其中肠道菌群中非常重要的Prevotella copri由于不在Refseq的完整测序基因组中,数据库没有包括,直接使用这个自带数据库会导致肠道菌群分析存在严重偏差,个别样本甚至95%以上都是该菌,如下面的这个样本:

可以看到红色箭头指向的Prevotella copri菌占比很高;假如数据库没有包含该菌注释,那么在后续分析的时候会错过一些重要信息。
谷禾数据分析使用了包括RefSeq 最近的99版本基因组数据,涵盖细菌、病毒、古菌、原生动物,不仅是完成的基因组还涵盖了基因组框架的物种。另外加入了IMG的真菌和细菌的基因组数据,以及真核寄生生物的数据库。
数据比对和统计分析结果如下,报告使用Pavian展示:

可以看到图中的红色框内,结果中能比对上数据库的比例大部分在90%以上,不能比上的只有不到10%的比例。
对应的物种构成表:

红色箭头指向的Max列,是后面列出样本reads数的总和。点击可以排序,简单方便。这个在线软件在谷禾报告中会给出相应的使用说明。
对应的每一个样的物种构成丰度:


以上的两张桑基图能将菌属构成,层级关系展示出来,更加直观。
接着看分析,下图是统计检验结果,共找到85个显著差异的菌,结果如下:

菌株部分发现42个差异菌株:

基于差异物种,使用spearmanCC,我们同样构建了精神病患者与健康对照两组差异菌的网络构成。

接着进一步分析了功能代谢,重点关注肠道菌群产生的神经递质和脑肠轴相关的代谢途径,也就是GBM模块与疾病分组和血清神经递质的关联关系。
前面已经提到,肠道菌群受年龄、饮食方式以及身体状况等影响,这些因素本身会导致肠道菌群状况产生偏差,如果不对这些因素进行统计控制,会对统计结果尤其是统计效力产生很大影响。
控制环境因素的影响
一般在统计上可以通过控制样本分组这些因素的分布,使其分组之间的因素基本一致或增加样本群体数量来增加统计效力。也可以通过统计方法如偏相关或加入协变量控制的GLM模型等方式来控制这些环境因素的影响。
这里的例子很好的显示了对这些因素控制和不做控制带来的统计分析结果的差异。
不 做 控 制
下图为不做控制的菌群总体GBM代谢模块与血清神经递质以及样本元数据的相关情况:

上图可以看到完全没有显著相关。
控 制 协 变 量
对性别、年龄以及食物摄入特点进行协变量控制之后的偏相关分析结果:

在这张图,可以找到显著的相关了。
可以看到有没有控制协变量对结果会造成影响,因此这里强调下协变量控制的重要性。
那么这里就意味着很多关联性被性别、年龄和饮食特点干扰了。
可以看到其中与是否患病的诊断(Diagnosis)相关的仅有DOPAC synthesis这一项,其他的大部分都不相关,该代谢通路同样在抑郁症人群中被发现存在显著相关,且与抑郁程度直接相关。
进一步的问题来了。
是否是菌群产生的神经递质直接影响了血清神经递质的量,并引发精神病呢?
我们发现整体菌群的主要神经递质代谢虽然部分与精神病患者异常的血清神经递质存在相关,但是关联性较弱,且差异并不明显。这就带来了一个思考:差异菌是否参与了这些神经递质的代谢异常。
我们仅分析了85个差异菌对应的GBM模块的代谢通路水平与血清神经递质的关联性(同样使用了偏相关):

除了与诊断分组全部强相关之外,与神经递质也有大量相关。但是当我们直接比对GBM模块代谢与相应神经递质的关系时发现,并不直接对应,也不存在单纯的菌群的合成途径升高对应血清神经递质的升高,甚至出现相反的情况。
差异菌可能是对神经递质敏感的菌
基于上面的结果,我们推测,这些差异菌实际上并不是直接导致神经递质异常的原因,很可能是对神经递质异常敏感的菌,当精神病患者出现特定神经递质水平异常时,会通过代谢或底物变化诱导这些菌的生长或抑制这些菌生长。
另一个证据是论文中提及这些患者中的一部分经过治疗或用药后3个月又进行了一次检测,其中一半以上的差异菌和健康对照相比都不再显著。

虽然没有直接找到菌群作为精神病发病因素的证据,但精神病患者确实存在特征菌群的变化。
那么这些特征变化的菌群是否能帮助我们对精神病进行诊断或区分呢?
使用随机森林,我们提取了最重要的10个菌作为区分特征,其分布如下:

ROC的结果如下:

没有达到论文中提及的89.56%的水平,但显示确实可以一定程度上区分。
需要注意的是,该研究中仅招募急性复发精神分裂症(ARSCZ)和首发精神分裂症(FESCZ)患者。该文中分析内容都是基于这个前提。因此我们认为对于部分用药后的精神分裂症患者菌群还有待研究。
另外使用热图决策树对所有元数据和菌群特征对精神病进行区分分析,发现仅使用血清色氨酸和MSCEIT量表两个指标就可以较好的区分精神病患者。
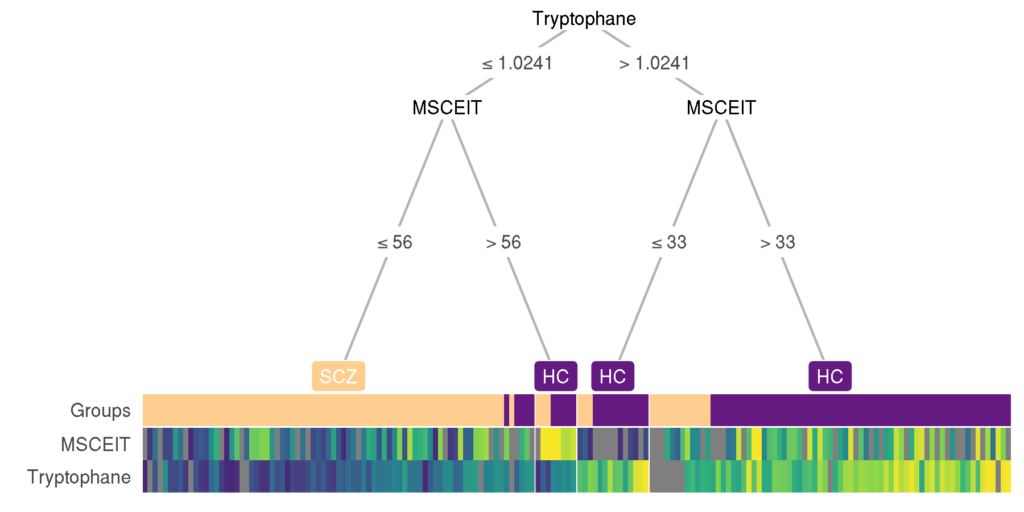
以上是我们结合自己构建的数据库,综合考虑多种因素,加入协变量控制分析得出的结果。
以上内容是对于该文章数据进行的分析。当然不同的文章,对应的分析可能会有些不同。
其他分析
以下图表为谷禾宏基因组分析网页报告的部分截取。谷禾宏基因组报告的形式在原先的基础上有所优化,采用网页报告的形式便于大家浏览,方便快捷。
bining分箱及评估
比如说想要拼出未知菌的基因组就要用到 bining分箱及评估:


基 因 预 测

物 种 分 析



功 能 分 析



测序深度的选择
目前宏基因组相较于16s,样本测序费用还是较高,除了建库费用主要原因来自于宏基因组样本测序量大,测序成本高相应分析成本也高(前面案例提到的文章测序数据量达到3000万reads,成本可想而知)。

除一些样本来源构成复杂的样本或者后续需要做分箱分析(主要获得样本里一些菌的基因组草图)的样本还有以基因序列和变异为目标的需要比较高的测序深度以外,大部分研究微生物群落以及其与环境互作等,其实不需要那么高的测序数据量。
下面我们从该案例文章170个样本中随机抽取100万reads做beta多样性PCA图。

左侧是使用了完整所有序列的PCA图,右侧是每个样本随机抽取100万reads的PCA图。
可以看到随机抽样100万reads之后的菌群分布情况和完整序列的完全相同。
既然100万reads数的菌群分布情况和完整序列没什么差异,我们也可以直接拿100万reads数进行后续分析。
而其他分析各个结果均显示在100万reads的基础上进行后续分析并没有影响最后的结论。
浅 宏 基 因 组
可能有人还没听过浅宏基因组,这里稍微介绍下。其实浅宏基因组跟宏基因组类似,最大的差别在于数据量。浅宏基因组测序深度相对较低(100万reads),但是物种的分辨率并没有低于一般宏基因组(5-6G)。
谷禾经过几个月的研发的测试,推出浅宏基因组测序分析服务,每个样本数据量不低于100万reads,不通过拼接组装,直接基于kraken2等kmer,或MetaPhlAn2等标记基因的参考基因组方法进行种属丰度分类。
结合其到菌株的物种分类和丰度数据可较16s方案下的PICRUST更加准确的预测基因构成。周期在:2-3周左右,尤其适合粪便样本,价格比16s测序价格稍高一点。
更高性价比,最大程度满足大家不同的需求。
有些同学可能还有疑惑,做这个真的可以发文章吗? 在你犹豫的时候,已经有浅宏基因组的文章发表了。
近日,美国梅奥诊所消化内科和肝病科 Purna C. Kashyap研究团队和明尼苏达大学生物科学学院 Dan Knights团队合作在 Cell 上发表了题为 Longitudinal Multi-omics Reveals Subset-Specific Mechanisms Underlying Irritable Bowel Syndrome 的文章。
该文章在菌群方面研究采用了宏基因组和16S,对粪便样本采用宏基因组,对黏膜样本采用16S,因为黏膜样本含有较高的人体DNA,16S更为合适。
粪便样本的宏基因组直接采用和RefSeq89版本进行比对注释,基因部分同时结合了序列比对和利用基因组数据直接提取注释相结合。
宏基因组测序能够提供菌株层面的分辨率,同时也是后续结构变异关联分析的必要条件,随着参考基因集的完善,中等测序深度的浅宏基因组将可以大量应用于这类研究中。
此外,对于这类文章的把握,实验方案的设计也很重要。
如何设计实验方案
这里我们提供一个谷禾参与设计的利用宏基因组技术,研究帕金森疾病与肠道菌群的研究方案示例。
帕金森与肠道菌群研究方案
帕金森氏病(PD)可能始于肠道中α-突触核蛋白原纤维的积聚,这可能与肠道营养不良有因果关系。
在帕金森氏病(PD)中,胃肠道功能很常见,通常先于运动征兆出现。PD可能是由病原体触发的肠道中起始,然后扩散到大脑。
已有多个人群队列研究显示PD患者人群和健康对照人群的肠道菌群Beta多样性存在显著差异。在Wallen等2020年的研究中发现有三个聚类的菌。
簇1由机会性病原体组成,所有PD均升高。
簇2是产生短链脂肪酸(SCFA)的细菌,PD均降低。
簇3是碳水化合物代谢的益生菌,并且PD升高。消炎产生SCFA的细菌的消耗和益生菌水平的升高是确定的。

目前在开展的帕金森队列研究中以荷兰DUPARC前瞻性队列研究为代表,临床试验注册于2019.11.28日。
研 究 方 案
招募150名从头开始研究PD的受试者。参加者将在1年和3年后接受随访评估,以期每3年进行一次扩展随访。
受试者具有广泛的特征,可以主要评估PD的三个主要领域内的目标:认知,胃肠功能和视觉。
这包括
脑磁共振成像(MRI);
脑胆碱能PET显像与氟乙氧基苯甲酸(FEOBV-PET);
具有氟多巴的脑多巴胺能PET成像(FDOPA-PET);
详细的神经心理学评估,涵盖所有认知领域;肠道微生物组组成;
肠壁通透性光学相干断层扫描(OCT);
基因分型运动和非运动症状;
总体临床状况和生活方式因素,包括饮食评估;
血液和粪便的储存,用于进一步分析炎症和代谢参数。
DUPARC是第一个在一段时间内将数据合并但不限于PD受试者认知,胃肠功能和视力的非运动领域数据的研究。作为一个从头开始的PD队列研究,以未接受过治疗的受试者作为基础,DUPARC为生物标志物的发现和验证提供了独特的机会,而不会造成多巴胺能药物的混杂影响。

现有的研究已经明确发现,多巴胺能药物会对菌群和代谢造成干扰,此外如儿茶酚-O-甲基转移酶抑制剂会显著增加乳杆菌科的含量。
另一项关联研究显示儿茶酚-O-甲基转移酶抑制剂(P = 4E-4),抗胆碱能药(P = 5E-3)和可能的卡比多巴-左旋多巴(P = 0.05)均对肠道菌群产生独立显著影响。
因此在构建研究队列时需要区分接受过治疗和未接受过治疗患者,对于接受治疗患者需要采集完整的用药记录信息。
研 究 方 向
目前帕金森与肠道菌群的研究有两种研究方向:
01
向菌群代谢产物,免疫信号,神经递质等方向探索
进一步深入解析肠道菌群在帕金森疾病进展和形成过程中扮演的角色,从已有的菌群构成向菌群代谢产物,免疫信号以及神经递质等方向探索。
单纯的纳入100例左右患者与相应对照人群检测菌群构成已有较多研究,只能从中国人群方面提供人群特异性变化,预期应该会有显著差异,但高水平研究论文较为困难。
研究方法上的改进可以纳入更加完善的临床和生理指标,如药物、认知,胃肠功能和视觉,以及更细化的脑磁共振成像(MRI)和代谢组特征进行组合统计分析。
另外可以从16S改为使用宏基因组测序,从更深和菌株层面发现关键群体以及基因关联。样本人群可以在100例左右,如果有可能尽量选择多中心或多个时间点,作为独立验证队列,以提高研究可信度。
02
解决现有治疗和疾病进展的个体化差异与菌群之间的关联和机制
另一个方向是从药物和疾病进展评估角度,这个方向需要从持续的样本收集和追踪,并纳入更多的临床和治疗信息,从而解决现有治疗和疾病进展的个体化差异与菌群之间的关联和机制。
这类研究纳入患者数量可以在50~100例之间,以病人为主,但需要持续跟踪,研究方案可以选择16S或宏基因组。
上述两个方向可根据临床患者招募和临床条件自行选择或组合。如果对照人群较难招募,建议以第二方向为主。
以上信息与大家分享交流,希望可以对即将或正在开展宏基因组研究的同仁们有所帮助。技术在不断进步,有价值的东西变得更好的方法就是不断更新迭代它,欢迎大家留言和交流。

