- 1第七章:使用Netlify零成本部署组件文档_netlify github教程
- 2SSH 解析 | 关键参数 | 安全配置_ssh配置步骤
- 3win11系统提示“无法成功完成操作,文件包含病毒”的原因和解决方法_win11提示无法成功完成操作
- 4【Linux命令详解 - ssh命令】 ssh命令用于远程登录到其他计算机,实现安全的远程管理_用ssh登录其他电脑
- 5elasticsearch 常用命令_elasticsearch 命令
- 6php 访问 github,一篇文章搞定Github API 调用 (v3)
- 7【C++】BFS类型题目总结_c++与门
- 8【Linux】开始认识软硬链接
- 9面试 Python 基础八股文十问十答第七期
- 10基于MATLAB的神经网络进行手写体数字识别(含鼠绘GUI / 数据集:MNIST)_matlab mnist
SCS【9】单细胞转录组之构建细胞轨迹 (Monocle 3)_monocle3 指定起点
赞
踩

点击关注,桓峰基因
桓峰基因公众号推出单细胞系列教程,有需要生信分析的老师可以联系我们!首选看下转录分析教程整理如下:
SCS【4】单细胞转录组数据可视化分析 (Seurat 4.0)
SCS【6】单细胞转录组之细胞类型自动注释 (SingleR)
SCS【7】单细胞转录组之轨迹分析 (Monocle 3) 聚类、分类和计数细胞
SCS【8】单细胞转录组之筛选标记基因 (Monocle 3)
SCS【9】单细胞转录组之构建细胞轨迹 (Monocle 3)
今天来说说单细胞转录组数据的细胞轨迹分析,学会这些分析结果,距离发文章就只差样本的选择了,有创新性的样本将成为文章的亮点,并不是分析内容了!
这期继续介绍 Monocle 3 软件包用于研究细胞拟时性分析也就是细胞的生长发育轨迹分析。
前 言
单细胞转录组测序(scRNA-seq)实验使我们能够发现新的细胞类型,并帮助我们了解它们是如何在发育过程中产生的。Monocle 3包提供了一个分析单细胞基因表达实验的工具包。
Monocle 3可以执行三种主要类型的分析:
-
聚类、分类和计数细胞。单细胞RNA-Seq实验允许发现新的(可能是罕见的)细胞亚型。
-
构建单细胞轨迹。在发育、疾病和整个生命过程中,细胞从一种状态过渡到另一种状态。Monocle 3可以发现这些转变。
-
差异表达分析。对新细胞类型和状态的描述,首先要与其他更容易理解的细胞进行比较。Monocle 3包括一个复杂的,但易于使用的表达系统。
工作流程图如下:
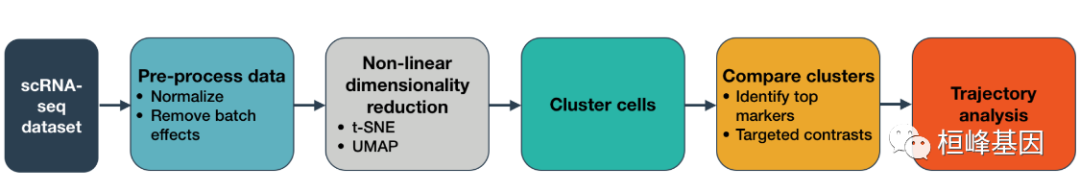
也就是流程图中的橙色框框的部分,包括构建单细胞轨迹。
在发育过程中,为了对刺激作出反应,在整个生命过程中,细胞从一种功能“状态”过渡到另一种功能“状态”。处于不同状态的细胞表达不同的基因集,产生一组动态的蛋白质和代谢物来完成它们的工作。当细胞在不同状态间移动时,它们会经历一个转录重新配置的过程,其中一些基因被沉默,另一些基因被激活。这些短暂的状态通常很难描述,因为在更稳定的端点状态之间净化细胞可能是困难的或不可能的。单细胞RNA-Seq可以让你看到这些状态,而不需要纯化。然而,要做到这一点,我们必须确定每个细胞在可能状态的范围内的位置。
Monocle介绍了使用RNA-Seq进行单细胞轨迹分析的策略。Monocle 不是通过实验将细胞纯化成离散状态,而是使用一种算法来了解每个细胞作为动态生物过程的一部分必须经历的基因表达变化的顺序。一旦了解了基因表达变化的整体“轨迹”,Monocle 就可以将每个细胞放在轨迹中适当的位置。然后,您可以使用 Monocle 的差异分析工具包来查找在轨迹过程中受调控的基因,如寻找作为伪时间函数变化的基因一节中所述。如果过程有多个结果,Monocle 将重建一个“分支”轨迹。这些分支对应着细胞的“决策”,Monocle 提供了强大的工具来识别受这些分支影响的基因和参与产生这些分支的基因。
数据读取
重构轨迹的工作流程非常类似于聚类的工作流程,但是有一些额外的步骤。为了说明工作流程,我们将使用另一个秀丽隐杆线虫数据集,这个数据集来自Packer和Zhu等人。他们的研究包括整个发育中的胚胎的时间序列分析。我们将检验数据的一小部分,其中包括大部分神经元。
expression_matrix <- readRDS("packer_embryo_expression.rds")
cell_metadata <- readRDS("packer_embryo_colData.rds")
gene_annotation <- readRDS("packer_embryo_rowData.rds")
cds <- new_cell_data_set(expression_matrix, cell_metadata = cell_metadata, gene_metadata = gene_annotation)
- 1
- 2
- 3
- 4
- 5
- 6
Pre-process the data
预处理的工作原理与聚类分析完全相同。这一次,我们将使用不同的策略进行批量校正,这包括Packer和Zhu等人在他们最初的分析中所做的:
cds <- preprocess_cds(cds, num_dim = 50)
cds <- align_cds(cds, alignment_group = "batch", residual_model_formula_str = "~ bg.300.loading + bg.400.loading + bg.500.1.loading + bg.500.2.loading + bg.r17.loading + bg.b01.loading + bg.b02.loading")
- 1
- 2
- 3
注意,除了使用 align_cds() 的 alignment_group 参数(用于对齐单细胞组)之外,我们还使用了 residual_model_formula_str 。这个目的是为了减去连续的影响。你可以用它来控制每个细胞中线粒体读取的分数,这有时被用作每个细胞的质量控制指标。在这个实验中(和许多 scRNA-seq 实验一样),一些细胞自发裂解,在加载到单细胞文库准备之前立即将它们的 mRNA 释放到细胞悬液中。这种“上清 RNA ”在一定程度上污染了每个细胞的转录组。幸运的是,可以相当直接地估计每批细胞的背景污染水平并将其减去,这就是Packer等人在最初的研究中所做的。每一列bg.300。装载时,bg.400。加载,对应一个背景信号池可能被污染。在 residual_model_formula_str 中传递这些列作为项,告诉 align_cds() 在降维、聚类和轨迹推断之前减去这些信号。注意,您可以使用 alignment_group、residual_model_formula 或两者调用align_cds()。
Reduce dimensionality and visualize the results
接下来,我们降低数据的维数。然而,与聚类不同的是,UMAP 和 t-SNE 都能很好地工作,在这里我们强烈建议您使用UMAP,默认方法:
cds <- reduce_dimension(cds)
plot_cells(cds, label_groups_by_cluster = FALSE, color_cells_by = "cell.type")
- 1
- 2
- 3
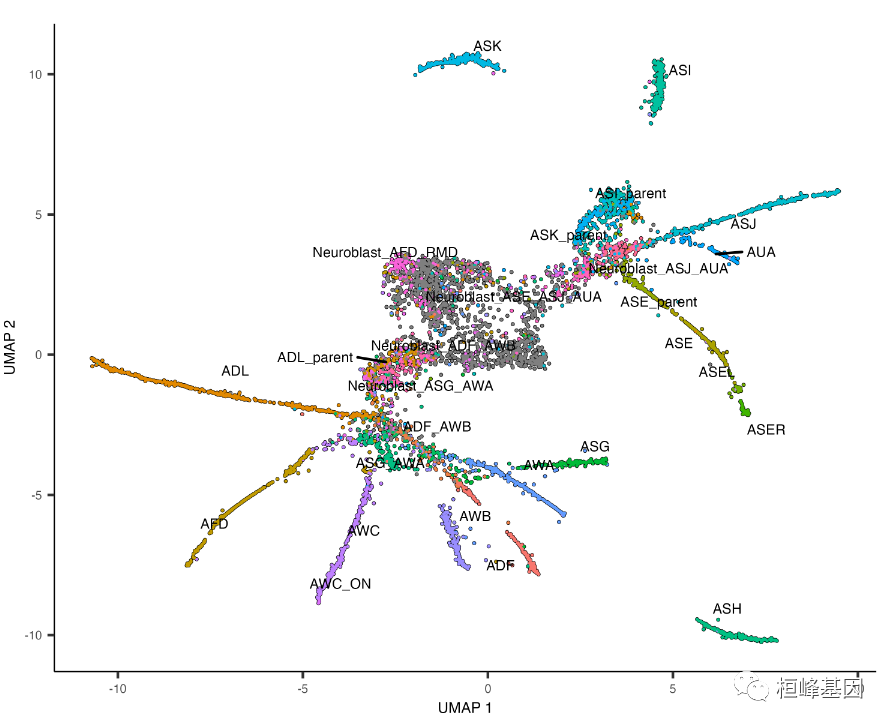
正如您所看到的,尽管我们只关注这个数据集的一小部分,Monocle 还是重建了一个具有众多分支的轨迹。在UMAP上叠加手动注释可以发现,这些分支主要由一个细胞类型所占据。
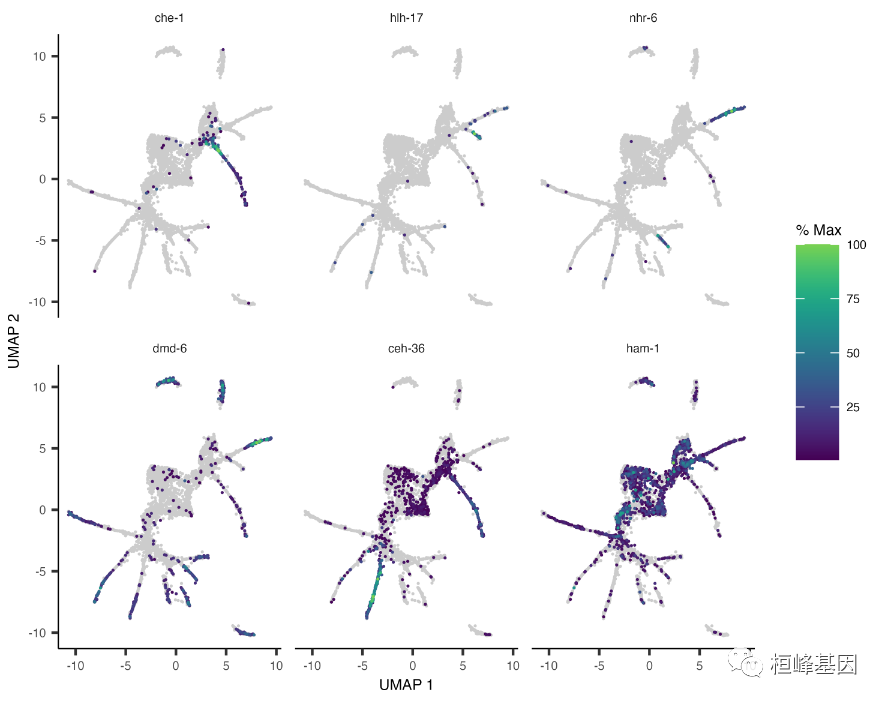
与聚类分析一样,可以使用 plot_cells() 来可视化单个基因沿轨迹的变化情况。让我们来看看一些在纤毛神经元中具有有趣表达模式的基因:
ciliated_genes <- c("che-1", "hlh-17", "nhr-6", "dmd-6", "ceh-36", "ham-1")
plot_cells(cds, genes = ciliated_genes, label_cell_groups = FALSE, show_trajectory_graph = FALSE)
- 1
- 2
- 3
- 4
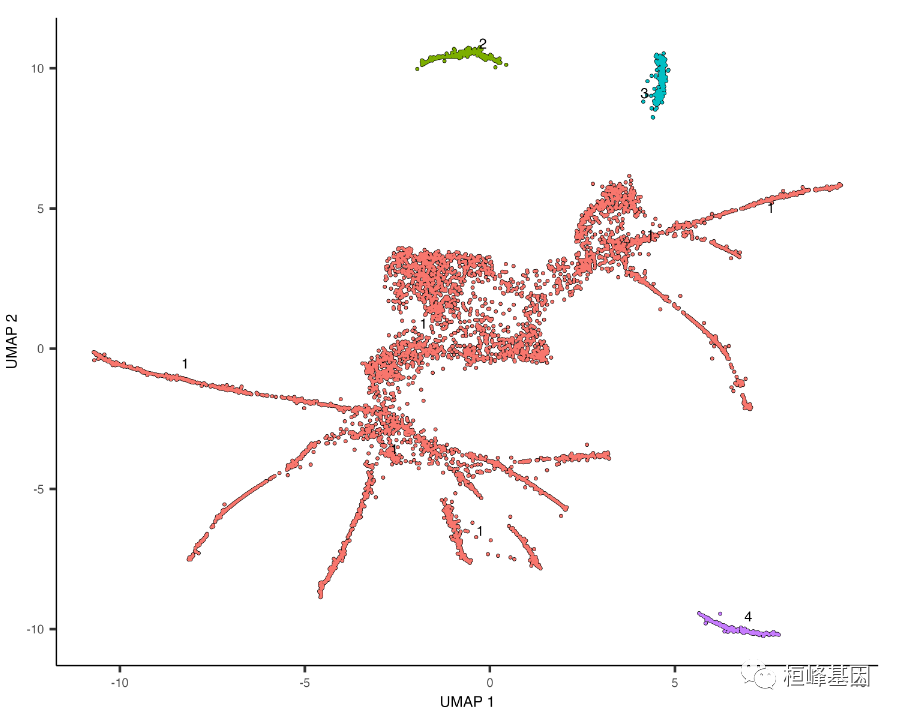
细胞聚类
尽管细胞可以连续地从一种状态过渡到另一种状态,它们之间没有离散的边界,Monocle并不假设数据集中的所有细胞都来自一个共同的转录“祖先”。在许多实验中,实际上可能有多个不同的轨迹。例如,在一个对感染有反应的组织中,组织内的免疫细胞和基质细胞会有非常不同的初始转录组,对感染的反应也会非常不同,所以应该是同一轨迹的一部分。
Monocle 能够通过聚类过程学习何时细胞应该放置在相同的轨迹,而不是单独的轨迹。回想一下,我们运行 cluster_cells() 时,每个细胞不仅被分配给一个簇,还被分配给一个分区。当你学习轨迹的时候,每个分区最终会变成一个单独的轨迹。我们像以前一样运行 cluster_cells()。
cds <- cluster_cells(cds)
plot_cells(cds, color_cells_by = "partition")
- 1
- 2
- 3
学习轨迹
接下来,我们将使用learn_graph()函数在每个分区中拟合一个主图,该图将用于许多下游步骤,如分支分析和微分表达式。
cds <- learn_graph(cds)
plot_cells(cds, color_cells_by = "cell.type", label_groups_by_cluster = FALSE, label_leaves = FALSE,
label_branch_points = FALSE)
- 1
- 2
- 3
- 4
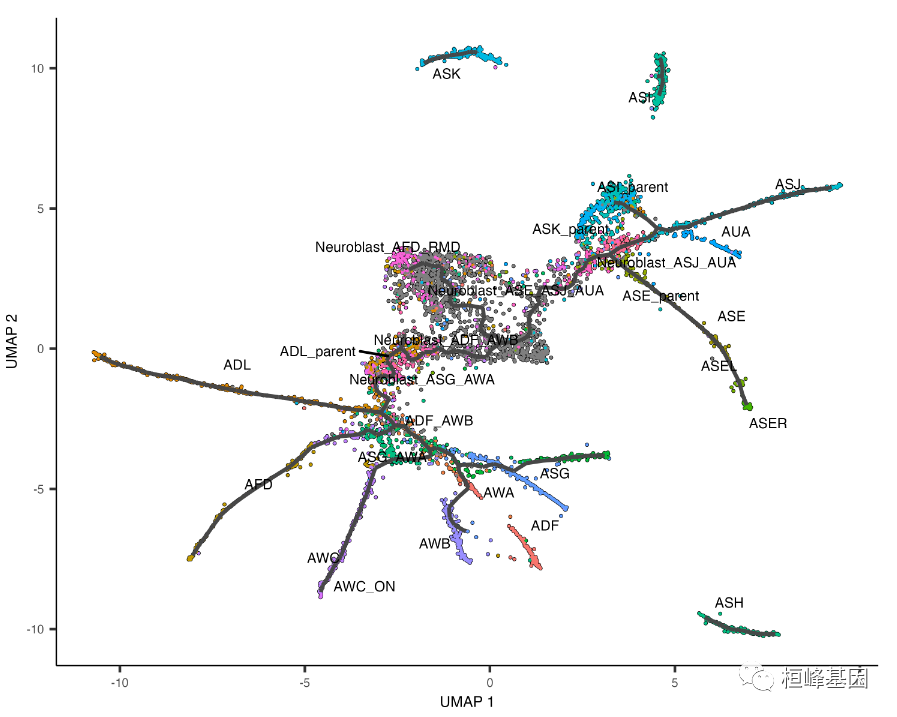
按拟时间顺序排列细胞
一旦我们掌握了一个图表,我们就可以根据细胞在发育过程中的进展来对它们进行排序。Monocle 用拟时间来测量这个过程。下面的方框定义了拟时间。拟时间是什么?

为了按顺序排列细胞,我们需要告诉 Monocle 生物过程的“起点”在哪里。为此,我们在图中选择标记为轨迹“根”的区域。在时间序列实验中,这通常可以通过在 UMAP 空间中找到早期时间点的细胞所占据的点来实现:
plot_cells(cds, color_cells_by = "embryo.time.bin", label_cell_groups = FALSE, label_leaves = TRUE,
label_branch_points = TRUE, graph_label_size = 1.5)
- 1
- 2
- 3
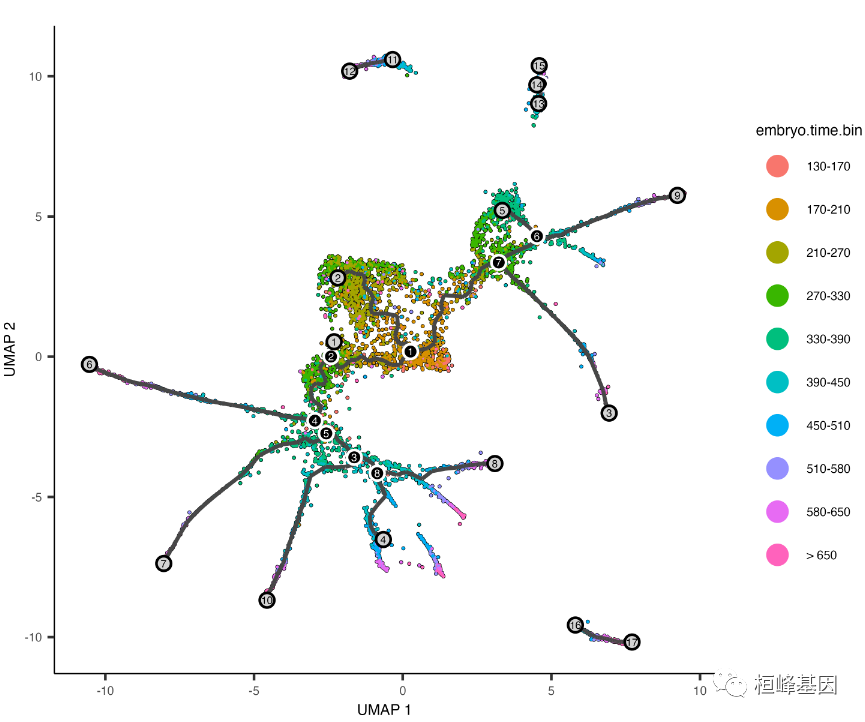
黑线表示图表的结构。注意,图不是完全连接的:不同分区中的细胞位于图的不同组件中。带数字的圆表示图中的特殊点。每个叶,用浅灰色圆圈表示,对应于轨迹的不同结果(即细胞命运)。黑圈表示分支节点,在分支节点中细胞可以移动到几种结果中的一种。您可以通过 plot_cells 的label_leaves 和 label_branch_points 参数来控制这些参数是否显示在绘图中。请注意,圆圈内的数字只供参考用途。
现在我们已经知道了早期细胞的位置,我们可以调用 order_cells(),它将计算每个细胞在拟时间中的位置。为了做到这一点,order_cells() 需要指定轨迹图的根节点。如果不将它们作为参数提供,将启动一个图形用户界面,用于选择一个或多个根节点。
cds <- order_cells(cds)
- 1
- 2

在上面的例子中,我们只选择了一个位置,但是您可以选择任意多个位置。用拟时间绘制细胞并给它们上色,可以看出它们是如何排序的:
plot_cells(cds, color_cells_by = "pseudotime", label_cell_groups = FALSE, label_leaves = FALSE,
label_branch_points = FALSE, graph_label_size = 1.5)
- 1
- 2
- 3
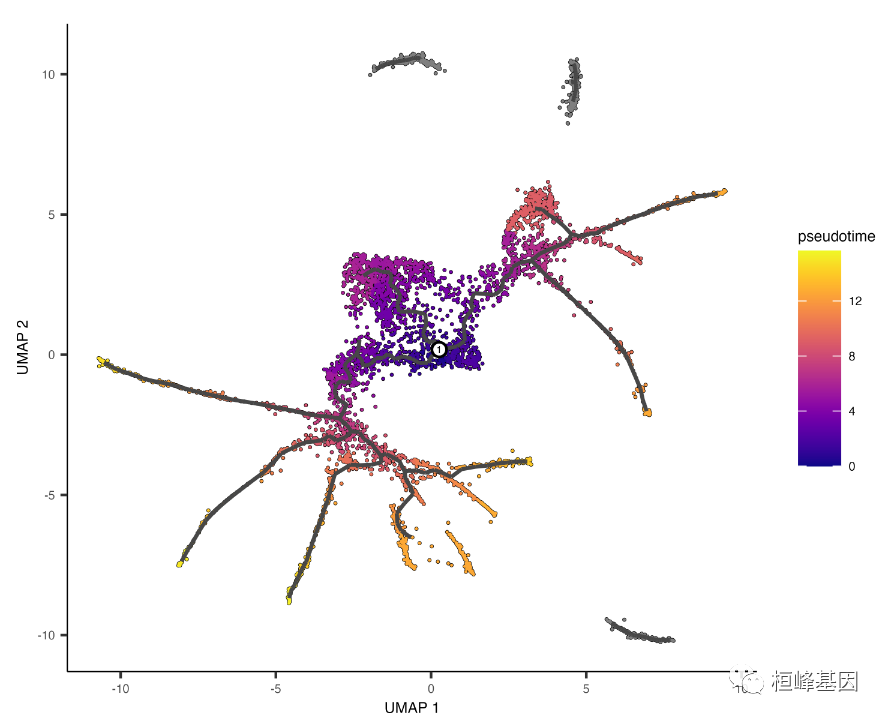
注意,有些细胞是灰色的。这意味着它们有无限的拟时间,因为从所选择的根节点无法访问它们。通常,分区上缺乏根节点的任何细胞都将被分配一个无限的拟时间。通常,每个分区至少应该选择一个根。
通常需要以编程方式指定轨迹的根,而不是手动选择它。下面的函数首先根据最接近的轨迹图节点对细胞进行分组。然后,计算每个节点上来自最早时间点的细胞的比例。然后,它选择早期细胞占用最多的节点,并将其作为根节点返回。
# a helper function to identify the root principal points:
get_earliest_principal_node <- function(cds, time_bin = "130-170") {
cell_ids <- which(colData(cds)[, "embryo.time.bin"] == time_bin)
closest_vertex <- cds@principal_graph_aux[["UMAP"]]$pr_graph_cell_proj_closest_vertex
closest_vertex <- as.matrix(closest_vertex[colnames(cds), ])
root_pr_nodes <- igraph::V(principal_graph(cds)[["UMAP"]])$name[as.numeric(names(which.max(table(closest_vertex[cell_ids,
]))))]
root_pr_nodes
}
cds <- order_cells(cds, root_pr_nodes = get_earliest_principal_node(cds))
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
通过 root_pr_node 参数将程序选择的根节点传递给 order_cells() 得到:
plot_cells(cds, color_cells_by = "pseudotime", label_cell_groups = FALSE, label_leaves = FALSE,
label_branch_points = FALSE, graph_label_size = 1.5)
- 1
- 2
- 3
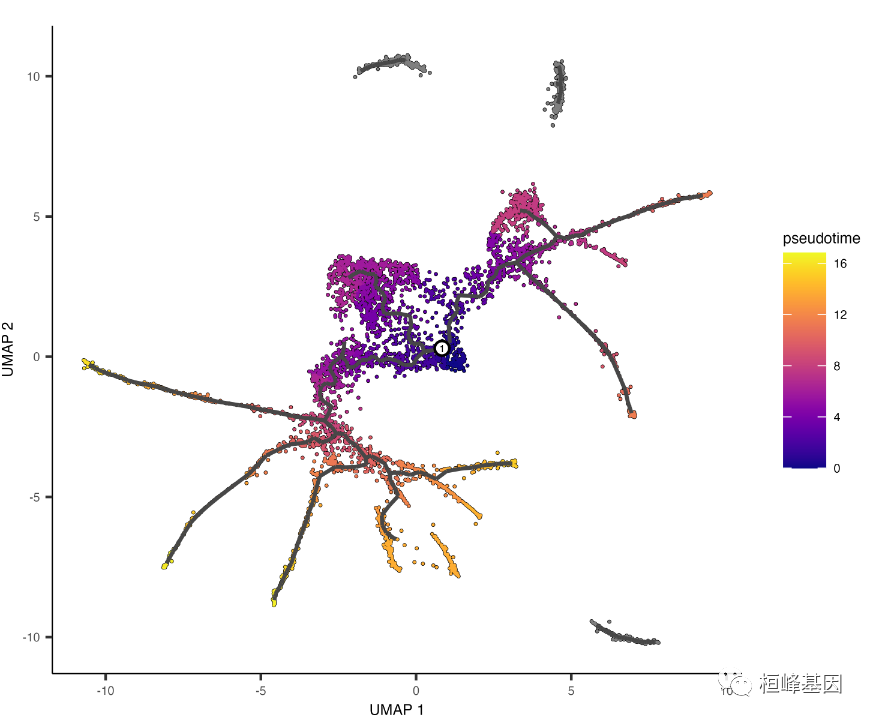
注意,通过使用 partitions() 函数首先按分区对细胞进行分组,我们可以很容易地在每个分区上实现这一点。这将导致所有细胞被分配一个有限的拟时间。
按分支筛选单细胞
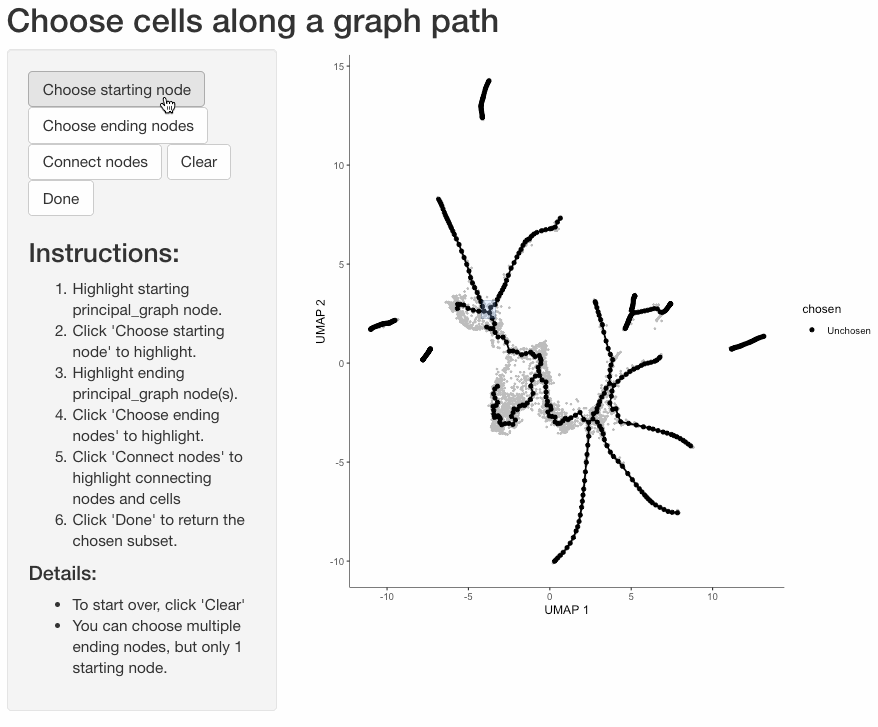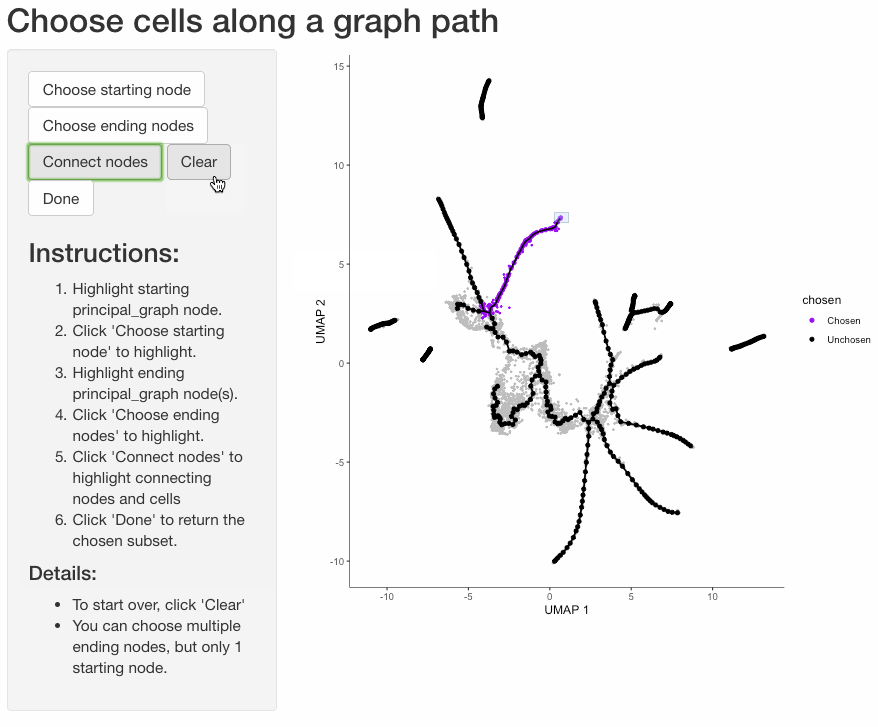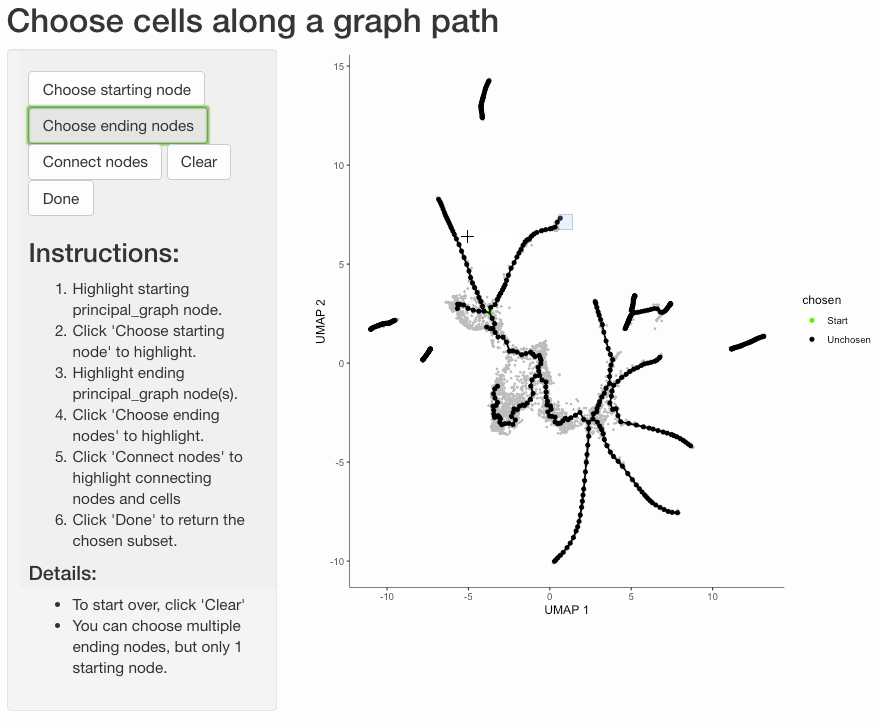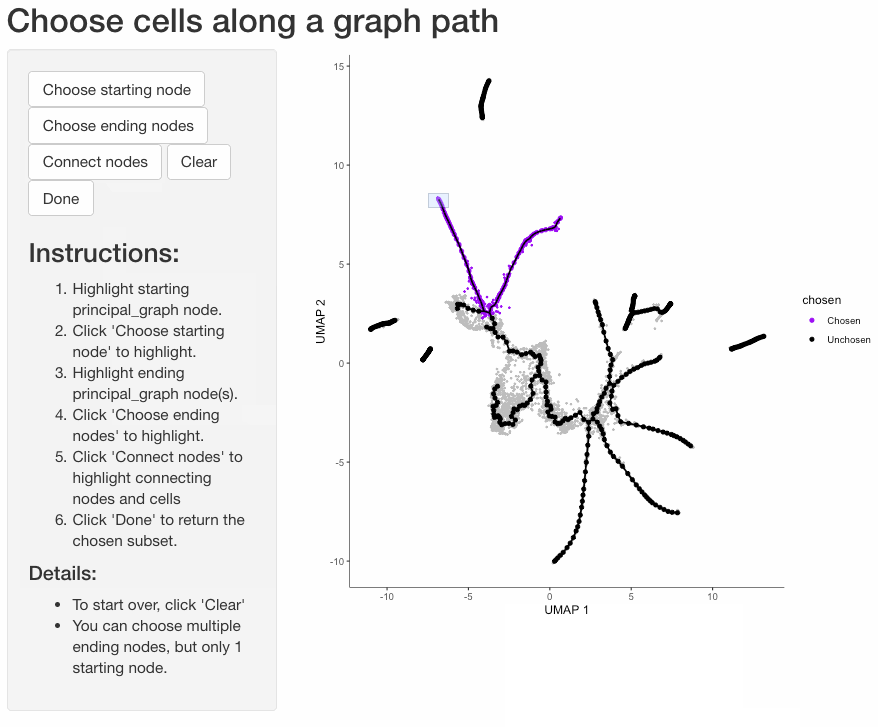
根据细胞在轨迹中的分支对细胞进行分组通常是有用的。函数 choose_graph_segments 允许您以交互方式这样做。
cds_sub <- choose_graph_segments(cds)
- 1
- 2
使用3D轨迹```
cds_3d <- reduce_dimension(cds, max_components = 3)
cds_3d <- cluster_cells(cds_3d)
cds_3d <- learn_graph(cds_3d)
cds_3d <- order_cells(cds_3d, root_pr_nodes = get_earliest_principal_node(cds))
cds_3d_plot_obj <- plot_cells_3d(cds_3d, color_cells_by = "partition")
- 1
- 2
- 3
- 4
- 5
- 6
- 7
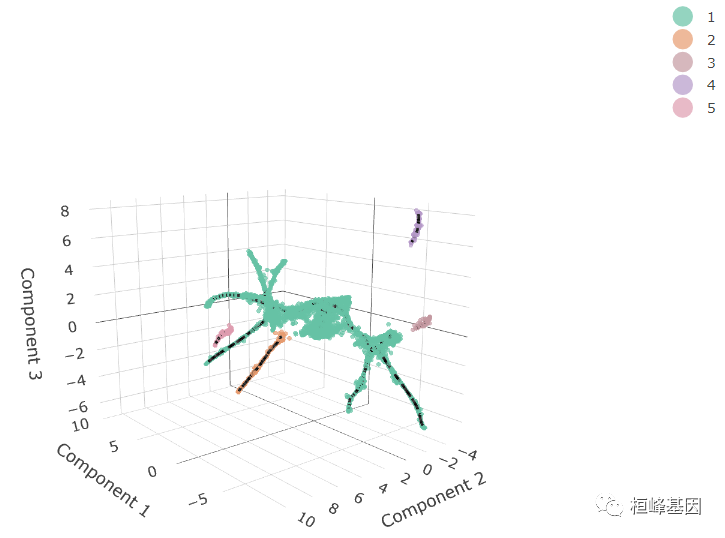
我们这期先分析第三部分,内容过多,一次完成有点太乱了,目前单细胞测序的费用也在降低,单细胞系列可算是目前的测序神器,有这方面需求的老师,联系桓峰基因,提供最高端的科研服务!
桓峰基因,铸造成功的您!
未来桓峰基因公众号将不间断的推出单细胞系列生信分析教程,
敬请期待!!
有想进生信交流群的老师可以扫最后一个二维码加微信,备注“单位+姓名+目的”,有些想发广告的就免打扰吧,还得费力气把你踢出去!
References:
-
Trapnell C. et. al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381-386 (2014).
-
Qiu, X. et. al. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979-982 (2017).
-
Cao, J. et. al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496-502 (2019).
-
Haghverdi, L. et. al. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36, 421-427 (2018).
-
McInnes, L., Healy, J. & Melville, J. UMAP: Uniform Manifold Approximation and Projection for dimension reduction. Preprint at https://arxiv.org/abs/1802.03426 (2018).
-
Traag, V.A., Waltman, L. & van Eck, N.J. From Louvain to Leiden: guaranteeing well-connected communities. Scientific Reportsvolume 9, Article number: 5233 (2019).
-
Levine, J. H. et. al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 162, 184-197 (2015).
-
Levine, J. H., et. al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 162, 184-197 (2015).