
- 1决策树_info[2,3]= bits
- 2Python(Cluster)_python cluster
- 32023HW红队作战工具
- 4IKEv2协议协商流程: (IKE-SA-INIT 交换)第一包_ikev2协商过程
- 5华为OD机试 - 字符串变换最小字符串(Java & JS & Python & C & C++)_华为od机试交换字符、字符串变换最小字符串
- 6Linux系统Ubuntu下安装Docker_ubuntu 版本 linux安装docker
- 7基于springboot校园志愿者管理系统系统、志愿者管理系统,附源码+数据库+论文+PPT,适合课程设计、毕业设计_志愿者系统设计
- 8一文全面盘点算法工程师大模型微调技能图谱
- 9一个人好几年没工作了怎么让他想去工作
- 10【2024华为OD机试真题C卷D卷-100分】悄悄话花费的时间【C++/Java/Python】_二叉树传递悄悄话
【第十一届泰迪杯数据挖掘挑战赛】A 题:新冠疫情防控数据的分析 思路+代码(持续更新)_泰迪杯新冠疫情数据分析
赞
踩
问题背景
自 2019 年底至今,全国各地陆续出现不同程度的新冠病毒感染疫情,如何控制疫情蔓
延、维持社会生活及经济秩序的正常运行是疫情防控的重要课题。大数据分析为疫情的精准
防控提供了高效处置、方便快捷的工具,特别是在人员的分类管理、传播途径追踪、疫情研
判等工作中起到了重要作用,为卫生防疫部门的管理决策提供了可靠依据。疫情数据主要包
括人员信息、场所信息、个人自查上报信息、场所码扫码信息、核酸采样检测信息、疫苗接
种信息等。
本赛题提供了某市新冠疫情防疫系统的相关数据信息,请根据这些数据信息进行综合分
析,主要任务包括数据仓库设计、疫情传播途径追踪、传播指数估计及疫情趋势研判等。
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
解决问题
- 根据核酸检测中阳性人员的出行时间与场所追踪密接者,将结果保存到
“result1.csv”文件中(文件模板见附件 1 中的 result1.csv)。 - 由问题 1 的结果,根据密接者的出行时间与场所追踪相应的次密接者,将结果保存
到“result2.csv”文件中(文件模板见附件 1 中的 result2.csv)。 - 建立模型,分析接种疫苗对病毒传播指数的影响。
- 根据阳性人员的数量及辐射范围,分析确定需要重点管控的场所。
- 为了更精准地进行疫情防控和人员管理,你认为还需要收集哪些相关数据。基于这
些数据构建模型,分析其精准防控的效果。
注 在解决上述问题时,要求结合赛题提供的数据信息表建立数据仓库,实现数据治理
的内容,请在论文中明确阐述做了哪些数据治理工作,具体是如何实现的。
!!注意:以下代码是在Aistudio上面写的,因此就没有建立相关数据库,根据题目要求你们自行建立数据库,然后在代码中进行读取就好了。
代码下载
代码下载地址:第十一届泰迪杯数据挖掘挑战赛-ABC-Baseline
-
大家Fork项目即可查阅所有代码了(free)
-
本项目仅供学习参考,鼓励大家以赛促学,为了保证比赛的公平性(只提供初级Baseline及简易思路分享)
-
若涉嫌违规,将会第一时间删除项目
注:思路仅代表作者个人见解,不一定正确。- 1
数据分析
- 导入常用的包
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import seaborn as sns
from tqdm import tqdm
import warnings
warnings.filterwarnings('ignore')
%matplotlib inline
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 导入文件的时候发现,附件的编码有问题,因此我们需要封装一个获取文件编码的函数
# 获取文件编码
import chardet
def detect_encoding(file_path):
with open(file_path,'rb') as f:
data = f.read()
result = chardet.detect(data)
return result['encoding']
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 读入所有附件
# 读取人员信息表
df_people = pd.read_csv('../datasets/附件2.csv',encoding = detect_encoding('../datasets/附件2.csv'))
# 读取场所信息表
df_place = pd.read_csv('../datasets/附件3.csv',encoding = detect_encoding('../datasets/附件3.csv'))
# 个人自查上报信息表
df_self_check = pd.read_csv('../datasets/附件4.csv',encoding = detect_encoding('../datasets/附件4.csv'))
# 场所码扫码信息表
df_scan = pd.read_csv('../datasets/附件5.csv',encoding = detect_encoding('../datasets/附件5.csv'))
# 核算采样检测信息表
df_nucleic_acid = pd.read_csv('../datasets/附件6.csv',encoding = detect_encoding('../datasets/附件6.csv'))
# 提交示例1
result = pd.read_csv('../datasets/result1.csv',encoding = detect_encoding('../datasets/result1.csv'))
# 提交示例2
result1 = pd.read_csv('../datasets/result2.csv',encoding = detect_encoding('../datasets/result2.csv'))
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 简单的查看一下提交示例
# 查看提交示例
result.head()
result1.head()
- 1
- 2
- 3
- 从提交示例可以看出,该问题应该是让我们制定一个策略去追踪密接者
- 并根据制定的策略获取密接者的其它信息
- 各附件的描述性统计
- 为了让描述性统计更直观,这里给描述性统计封装了一个函数
# 数据描述性统计 def summary_stats_table(data): ''' a function to summerize all types of data 分类型按列的数据分布与异常值统计 ''' # count of nulls # 空值数量 missing_counts = pd.DataFrame(data.isnull().sum()) missing_counts.columns = ['count_null'] # numeric column stats # 数值列数据分布统计 num_stats = data.select_dtypes(include=['int64','float64']).describe().loc[['count','min','max','25%','50%','75%']].transpose() num_stats['dtype'] = data.select_dtypes(include=['int64','float64']).dtypes.tolist() # non-numeric value stats # 非数值列数据分布统计 non_num_stats = data.select_dtypes(exclude=['int64','float64']).describe().transpose() non_num_stats['dtype'] = data.select_dtypes(exclude=['int64','float64']).dtypes.tolist() non_num_stats = non_num_stats.rename(columns={"first": "min", "last": "max"}) # merge all # 聚合结果 stats_merge = pd.concat([num_stats, non_num_stats], axis=0, join='outer', ignore_index=False, keys=None, levels=None, names=None, verify_integrity=False, copy=True, sort=False).fillna("").sort_values('dtype') column_order = ['dtype', 'count', 'count_null','unique','min','max','25%','50%','75%','top','freq'] summary_stats = pd.merge(stats_merge, missing_counts, left_index=True, right_index=True, sort=False)[column_order] return(summary_stats)

- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 人员信息表
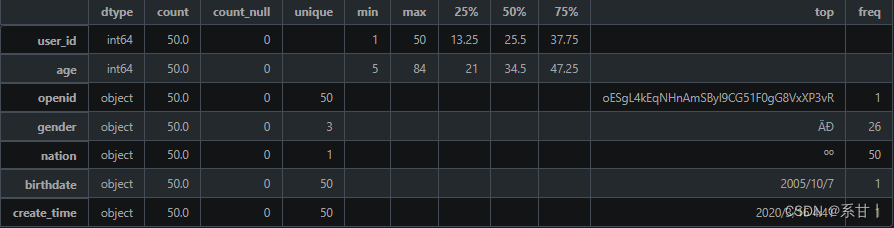
数据说明 - user_id:人员 ID,用于唯一标识一个人员。
- openid:微信 OpenID,用于关联该人员的微信账号信息。
- gender:人员的性别,可选值为“男”或“女”。
- nation:人员所属的民族,如汉族、蒙古族、藏族等。
- age:人员的年龄,以整数表示。
- birthdate:人员的出生日期,格式一般为“YYYY-MM-DD”。
- create_time:该记录的创建时间,用于记录人员信息的更新时间。
以下分析结果均基于示例数据
-
people总共50条数据。
-
年龄区间是[5,84]
Tips:年龄跨度比较大,自然而然,我们可以根据年龄做特征工程。
-
在gender中,总共有三个类别(可能存在“未知”类别),在题目中只给了两个类别。
Tips:如果后面需要根据性别进行分析或特征工程的话,需要考虑怎么处理第三个类别。
-
nation民族只有一个类别,而在全量数据中大概率不会只存在一个类别的
Tips:如果后续需要用到该列进行聚合分析或特征工程,可以在Baseline中写好动态的代码。
-
birthdate和create_time在这里都是对应着50个不一样的时间
Tips:注意关注时间的始末,与其它相关联的时间进行比较,这样可以挖掘出更多信息或筛选出一些异常情况。
在全量数据中,时间大概率是有重复值的,也要考虑重复时间是否对解题有一定的影响亦或者重复时间的含义。- 1
-
场所信息表
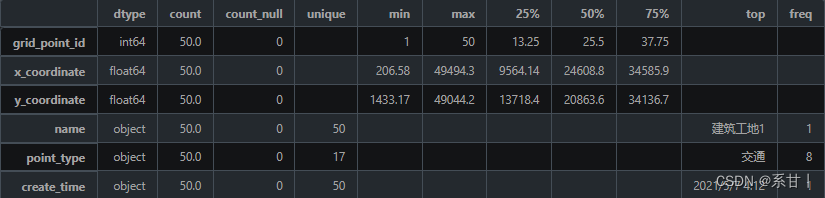
数据说明 -
grid_point_id:场所 ID,用于唯一标识一个场所。
-
name:场所的名称,如公司、餐厅、超市等。
-
point_type:场所的类型,如商业、娱乐、文化、医疗等。
-
x_coordinate:场所的 X 坐标,以米为单位,用于表示场所在地图上的位置。
-
y_coordinate:场所的 Y 坐标,以米为单位,用于表示场所在地图上的位置。
-
create_time:该记录的创建时间,用于记录场所信息的更新时间。
以下分析结果均基于示例数据
-
X、Y坐标,这或许是一个很好用来可视化的数据
Tips:可以根据X、Y坐标对其它特征进行可视化(包括但不限于name、point_type)
但是需要注意的是这只是示例数据,全量数据可能会比较庞大,可视化出来的效果可能没有理想那么好- 1
-
name是场所名,在示例数据中没有重复数据(但不代表全量数据中不会出现重复)
Tips:针对重复的场所名,是否可以聚合起来做数据统计呢?亦或者其它
-
point_type场所类型,在示例数据中总共有17个不同的场所类型,其中类型为娱乐的场所最多
Tips:娱乐只是在示例数据中的结果,不一定是全量数据的。可以根据这一列特征做更多的数据分析,或许还可以进行特征工程
全量数据中有可能出现不同样本中X,Y值相同而对应的name或point_type等其它特征不同的情况。具体问题具体分析,不要什么都当作异常值- 1
-
个人自查上报信息表
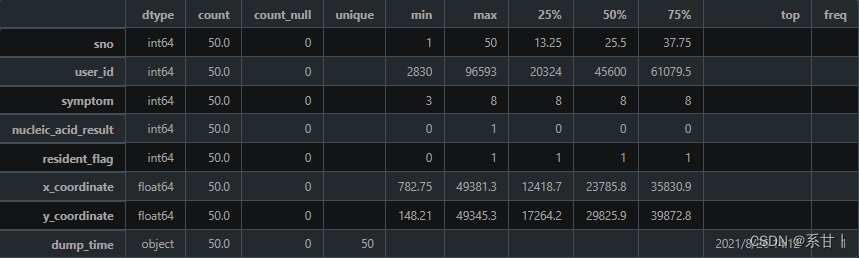
数据说明 -
sno:序列号,用于唯一标识一条自查记录。
-
user_id:人员 ID,对应于“人员信息表”中的 user_id,用于关联自查记录与相应的人员。
-
x_coordinate:上报地点的 X 坐标,以米为单位,用于表示上报地点在地图上的位置。
-
y_coordinate:上报地点的 Y 坐标,以米为单位,用于表示上报地点在地图上的位置。
-
symptom:症状,用于记录自查者的症状情况。可选值为:1 发热、2 乏力、3 干咳、4 鼻塞、5 流涕、6 腹泻、7 呼吸困难、8 无症状。
-
nucleic_acid_result:核酸检测结果,用于记录自查者的核酸检测情况。可选值为:0 阴性、1 阳性、2 未知(非必填)。
-
resident_flag:是否常住居民,用于记录自查者的居住情况。可选值为:0 未知、1 是、2 否。
-
dump_time:上报时间,用于记录自查记录的上报时间。
以下分析结果均基于示例数据
-
symptom,可以看出症状类别在示例数据中是不全的,在示例数据中几乎都是无症状(8)
Tips:在全量数据中,所有类别的数据应该都会存在的,因此在写Baseline的时候可以考虑先写好数据分析可视化的代码。
这一列特征还有一个特点是,在特征工程的时候,可以很好的和其它特征衍生出很多可解释性的交叉特征(eg:symptom-nucleic_acid_result)- 1
-
nucleic_acid_result,resident_flag同理
-
这里的X,Y坐标和上表的并不一样,可以挖掘一下两者的区别
Tips:可以根据这X,Y坐标确定该人在什么场所进行的信息上报。
-
dump_time(上报时间),可以将这一列和nucleic_acid_result,X、Y坐标结合,可以挖掘出阳性患者上报期间所在的场合以及周围的人
-
场所码扫码信息表
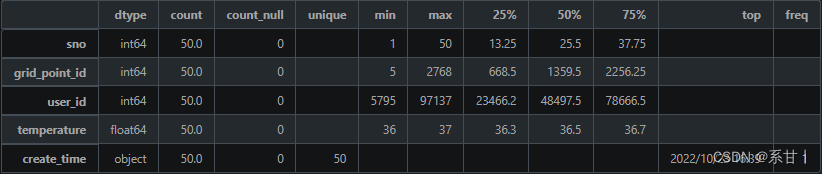
数据说明 -
sno:序列号,用于唯一标识一条扫码记录。
-
grid_point_id:场所 ID,对应于“场所信息表”中的 grid_point_id,用于关联扫码记录与相应的场所。
-
user_id:人员 ID,对应于“人员信息表”中的 user_id,用于关联扫码记录与相应的人员。
-
temperature:体温,用于记录扫码者的体温情况。
-
create_time:扫码记录时间,用于记录扫码记录的时间戳。
以下分析结果均基于示例数据
-
temperature(体温),在示例数据中最小值是36,最大值是37,这数值貌似都在人体正常体温的范畴
Tips:可以将该列与个人信息表中的特征进行交叉分析,在全量数据中大概率会有39左右或更高的体温,因此在写Baseline的时候最好将其考虑进去。
-
create_time(扫码记录时间),我们可以将扫码记录的时间当成该人员即时的体温时间,然后与其它表的特征及时间进行比较
Tips:例如可以与个人自查上报信息表的上报时间以及采样日期进行比较
-
核酸采样检测信息表
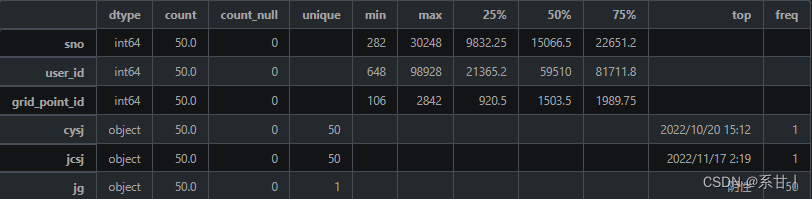
数据说明 -
sno:序列号,用于唯一标识一条核酸采样记录。
-
user_id:人员 ID,对应于“人员信息表”中的 user_id,用于关联核酸采样记录与相应的人员。
-
cysj:采样日期和时间,用于记录核酸采样的日期和时间。
-
jcsj:检测日期和时间,用于记录核酸检测的日期和时间。
-
jg:检测结果,用于记录核酸检测的结果。可选值为:阴性、阳性、未知。
-
grid_point_id:场所 ID,对应于“场所信息表”中的 grid_point_id,用于关联核酸采样记录与相应的场所。
以下分析结果均基于示例数据
-
这里出现了两个时间,一个是采样时间,一个是检测时间,那么按照逻辑来说检测时间是会比采样时间晚的
Tips:小心驶得万年船,我们在这里加个判断,如果判断成立,那么该样本就可以视为异常值了
-
对于结果这一列,在示例中结果均为阴性
Tips:我们知道它总共会是有三个类别的,因此写Baseline的时候尽量考虑进去
-
由于前两题没涉及到附件7,因此在这里就没有导入
-
数据可视化建议
数据分析时可以做以下可视化单表可视化
-
人员信息表:可以进行人口统计学分析,如性别、年龄、民族等分布情况,还可以通过人员 ID 与其他表格进行关联分析。
-
场所信息表:可以进行地理信息分析,如场所分布情况、场所类型分布情况、场所密度等分析。
-
个人自查上报信息表:可以进行疫情监测分析,如症状分布情况、症状与核酸检测结果的关联分析、上报人员的位置分布情况等分析。
-
场所码扫码信息表:可以进行疫情监测分析,如扫码记录分布情况、扫码记录与核酸检测结果的关联分析等。
-
核酸采样检测信息表:可以进行疫情监测分析,如阳性人员的分布情况、核酸检测阳性率分析、阳性人员的接触场所与密切接触者分析等。
关联分析
-
个人自查上报信息表和核酸采样检测信息表:可以分析个人上报的症状与核酸检测结果之间的关系,以及症状与检测结果对不同年龄、性别、民族等人群的影响。
-
场所信息表和场所码扫码信息表:可以分析不同场所的扫码情况,了解人们在哪些场所更容易扫码;也可以分析场所内体温异常者的情况,了解哪些场所的防疫工作存在漏洞。
-
个人自查上报信息表和场所码扫码信息表:可以根据个人自查上报的症状,分析不同场所的症状发生情况,了解哪些场所的防疫措施需要进一步加强。
-
核酸采样检测信息表和个人自查上报信息表、场所码扫码信息表:可以分析阳性人员的出行情况,追踪密接者,及时采取隔离措施。
-
Task1
Baseline实现了根据某个阳性人员的核酸检测记录,找出他在检测前后14天内去过的场所,然后再找出去过这些场所的人员,进而确定可能的密接者。具体的实现步骤如下:
-
首先,通过传入的阳性人员ID,在核酸检测记录中筛选出该阳性人员的检测记录,并获取阳性者的采样与检测时间。
-
接着,根据阳性人员在采样时的场所ID,确定第一个阳性人员所在的场所列表。
-
然后,通过阳性人员的ID与场所码扫码信息表进行拼接,获取阳性人员前后十四天所去的场所(第二个阳性人员所在的场所列表)。
-
将两个场所列表进行合并并去重。
-
最后,根据场所码扫码信息表中的所有User_id与场所信息表合并,通过场所列表和时间进行筛选,从而追踪密接者ID
Baseline实现了基于核酸检测记录,找出阳性人员在检测前后14天内去过的场所,并通过这些场所找出可能的密接者。
# 获取阳性者信息 positive_user_id = df_nucleic_acid[df_nucleic_acid['jg'] =='阳性']['user_id'].values.tolist() def Potential_contacts(df_people,df_place,df_self_check,df_scan,df_nucleic_acid,positive_user_id): # 筛选出阳性者的核酸检测记录 df_positive_test = df_nucleic_acid[df_nucleic_acid['user_id'] == positive_user_id] # 获取阳性者的检测时间 positive_test_time = pd.to_datetime(df_positive_test['cysj'].iloc[0]) df_self_check['dump_time'] = pd.to_datetime(df_self_check['dump_time']) df_scan['create_time'] = pd.to_datetime(df_scan['create_time']) # 获得阳性人员核酸检测的场所 positive_users_place1 = pd.merge(df_positive_test, df_place, on='grid_point_id')['name'].tolist() # 获得阳性人员在测验时间前后14天去的场所 positive_users_place2 = pd.merge(df_positive_test, df_scan, on='user_id')[['user_id','create_time','cysj','grid_point_id_y']] # 计算前14天和后14天 delta = pd.Timedelta(days=14) # 计算最小时间和最大时间 min_date = positive_users_place2['cysj'] - pd.Timedelta(days=14) max_date = positive_users_place2['cysj'] + pd.Timedelta(days=14) # 筛选出符合要求的数据 mask = (positive_users_place2['create_time'] >= min_date) & (positive_users_place2['create_time'] <= max_date) positive_users_place2 = positive_users_place2.loc[mask, ['user_id', 'grid_point_id_y']] positive_users_place2 = positive_users_place2.rename(columns={'grid_point_id_y': 'grid_point_id'}) positive_users_place2 = pd.merge(positive_users_place2, df_place, on='grid_point_id')['name'].tolist() # 将两个列表合并去重 positive_place = list(set(positive_users_place1+positive_users_place2)) # 获取去过上述场所的人员 # 按照密接时间筛选 df_potential_contacts = df_scan[(df_scan['create_time'] >= positive_test_time - pd.Timedelta('14D')) & (df_scan['create_time'] <= positive_test_time + pd.Timedelta('14D'))] # 按照场所筛选 df_potential_contacts = df_potential_contacts[df_potential_contacts['grid_point_id'].isin(df_place[df_place['name'].isin(positive_place)]['grid_point_id'])] # 整合信息并按照要求输出 result = pd.DataFrame({ '序号': range(1, len(df_potential_contacts)+1), '密接者ID': df_potential_contacts['user_id'].values, '密接日期': df_potential_contacts['create_time'].dt.date.astype(str), '密接场所ID': df_potential_contacts['grid_point_id'].values, '阳性人员ID': [positive_user_id] * len(df_potential_contacts) }) return result
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
为本题封装了名为 Potential_contacts的函数,该函数的目的是找到所有可能与阳性者有接触的人员信息。
函数的具体逻辑如下:
- 从 df_nucleic_acid 中获取 positive_user_id 对应的阳性者的核酸检测记录和检测时间。
- 将 df_self_check 和 df_scan 数据框中的时间列转换为 datetime 类型。
- 从 df_place 中获取 positive_user_id 在核酸检测时间点去过的场所列表 positive_users_place1。
- 从 df_scan 中获取 positive_user_id 在核酸检测时间点前后 14 天去过的场所列表 positive_users_place2。
- 将 positive_users_place1 和 positive_users_place2 合并去重得到 positive_place,即 positive_user_id 去过的所有场所。
- 从 df_scan 中筛选出在 positive_test_time 前后 14 天有扫码记录的人员(即潜在密接者)df_potential_contacts。
- 从 df_place 中筛选出 positive_place 中的场所,并将这些场所的 grid_point_id 与 df_potential_contacts 中的 grid_point_id 匹配得到所有潜在密接者的位置信息。
整合潜在密接者的信息和阳性者的信息,并返回一个数据框,其中包含序号、密接者 ID、密接日期、密接场所 ID 和阳性人员 ID 等信息。
Task2
def get_sub_contacts(df_potential_contacts, df_scan): # 修改列名,方便拼接 df_potential_contacts = df_potential_contacts.rename(columns = {'密接场所ID':'grid_point_id'}) # 筛选出所有在密接者场所出入过的UserID contacts = pd.merge(df_potential_contacts,df_scan,on='grid_point_id') # 去除密接者ID contacts = contacts.drop(contacts[contacts['密接者ID'] == contacts['user_id']].index) # 计算前后半个小时 delta = pd.Timedelta(minutes=30) # 筛选出次密接者 mask = (contacts['create_time'] >= contacts['密接日期']-delta) & (contacts['create_time'] <= contacts['密接日期']+delta) contacts = contacts[mask] # 整合信息并按照要求输出 result = pd.DataFrame({ '序号': range(1, len(contacts)+1), '次密接者ID': contacts['user_id'].values, '次密接日期': contacts['create_time'].values, '次密接场所ID': contacts['grid_point_id'].values, '阳性人员ID': contacts['密接者ID'].values }) return result
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
该函数的具体逻辑如下:
- 将df_potential_contacts中的 "密接场所ID "列重命名为 “grid_point_id”。
- 合并df_potential_contacts和df_scan中的grid_point_id列,以找到所有曾与密接者去过同一地点的用户。
- 删除任何用户ID与密接者ID相匹配的行,因为我们不想包括自我接触。
- 在密接者日期周围设置一个30分钟的时间窗口,筛选出在这时间区间内与密接者接触的用户。
为了找到次密接者,我们需要了解密接者密接期间所在的地点和与之接触的人员。
- 这段代码的逻辑是基于一个假设:
- 如果两个人曾经在同一个时间段内出现在同一地点,那么他们可能会接触到彼此,从而增加了被感染的风险。
-
因此,该函数首先将两个表格 (df_potential_contacts 和 df_scan)合并,找到所有在与密接者相同的场所出现过的用户。
-
接下来,该函数将筛选出密接者密接时间的前后半小时内有过接触的用户。
具体地,该函数使用 Pandas 库中的 Timedelta 函数设置一个时间窗口,
然后将 create_time 列中的日期和时间与 密接日期 进行比较,筛选出在这个时间窗口内的接触记录。
-
最后,根据result格式输出答案
Task3
病毒传播指数可以根据现有的数据表格中的核酸采样检测信息表和场所码扫码信息表来计算。一种常用的方法是使用传染病流行病学中的基本再生数R0,它代表一个感染者平均会传染多少其他人。
首先,我们可以根据场所码扫码信息表中的数据计算每个场所的平均体温,并根据该平均体温和感染者的体温来确定感染概率。最后,我们可以使用基本再生数公式(R0 = 感染概率 × 平均接触人数)来计算病毒传播指数。
具体步骤如下:
-
根据场所码扫码信息表中的数据,确定每个场所的温度分布的平均值和标准偏差。
-
根据场所码扫码信息表中的数据,确定感染者的体温。
-
根据感染者的体温和场所的平均体温,计算感染概率,例如P = exp(-(38-37)2/2σ2),其中σ是体温分布的标准差。
-
根据密接者表中的数据,确定平均接触人数。
-
计算基本再生数R0 = 感染概率 × 平均接触人数。
将计算出的病毒传播指数与接种疫苗信息表中的数据进行比较,分析接种疫苗对病毒传播指数的影响。
- 第一步:确定每个场所的温度分布的平均值和标准偏差
df_scan['temperature_mean'] = df_scan.groupby('grid_point_id')['temperature'].transform('mean')
df_scan['temperature_std'] = df_scan.groupby('grid_point_id')['temperature'].transform('std')
- 1
- 2
- 第二步:根据场所码扫码信息表和核酸采样信息表,确定感染者的平均体温
df_positive = pd.merge(df_scan, df_nucleic_acid[df_nucleic_acid['jg'] == '阳性'][['user_id']], on='user_id', how='inner')
df_positive['temperature_mean_positive'] = df_positive.groupby('grid_point_id')['temperature'].transform('mean')
- 1
- 2
- 第三步:计算感染概率
df_positive['infection_prob'] = np.exp(-((df_positive['temperature_mean_positive'] - df_positive['temperature_mean']) ** 2) / (2 * df_positive['temperature_std'] ** 2))
- 1
- 第四步:根据密接者表中的数据,确定平均接触人数
# 根据阳性人员ID和密接场所ID进行分组,并统计每个组内密接者数量
grouped = result1.groupby(['阳性人员ID', '密接场所ID'])['密接者ID'].count().reset_index()
# 将统计结果返回到原数据集中
result1 = pd.merge(result1, grouped, on=['阳性人员ID', '密接场所ID'], how='left')
result1_count = result1.rename(columns={'密接者ID_y': '密接者数量'})
result1_count['平均接触人数'] = result1_count.groupby(['阳性人员ID', '密接场所ID'])['密接者数量'].transform('mean')
df_positive = pd.merge(result1_count, df_positive, left_on='阳性人员ID', right_on='user_id')
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 第五步:根据感染概率和每个感染者在每个场所的平均接触次数计算病毒传播指数。
df_positive['label'] = df_positive['infection_prob'] * df_positive['平均接触人数']
- 1
- 第六步:与疫苗接种信息表合并
# 疫苗接种信息表
df_vaccine_info = pd.read_csv('../datasets/附件7.csv',encoding = detect_encoding('../datasets/附件7.csv'))
df = pd.merge(df_vaccine_info, df_positive, on='user_id')
# 去除没有label的数据
df = df.dropna()
- 1
- 2
- 3
- 4
- 5
- 第七步:拟合数据,分析特征重要性
col = ['age', 'gender','inject_times', 'vaccine_type','label'] df = df[col] # 对类别列进行数值编码(你也可以用其它编码进行特征工程) from sklearn.preprocessing import LabelEncoder from sklearn.ensemble import RandomForestRegressor # 创建 LabelEncoder 对象 le = LabelEncoder() # 对 nject_times 和 vaccine_type 进行数值编码 df['nject_times'] = le.fit_transform(df['nject_times']) df['vaccine_type'] = le.fit_transform(df['vaccine_type']) # 创建随机森林回归模型 rf = RandomForestRegressor(n_estimators=100, random_state=2023) # 拟合数据 X= df.drop('label',axis=1) y = df['label'] rf.fit(X, y) # 得到特征重要性 importances = rf.feature_importances_ # 将特征重要性排序 indices = np.argsort(importances)[::-1] # 将特征名称按照重要性排序 names = [f'Feature {i}' for i in range(X.shape[1])] sorted_names = [names[i] for i in indices] # 绘制特征重要性柱状图 plt.figure() plt.title("Feature Importance") plt.bar(range(X.shape[1]), importances[indices]) plt.xticks(range(X.shape[1]), sorted_names, rotation=90) plt.show()
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
Task3的方案属于抛砖引玉,参考思路即可
-
在该方案中,有很多因素并没考虑进去,建模的时候只考虑了年龄、性别、接种疫苗的类别
-
在该方案中,有很多决策是可以优化的,例如每个场所的温度分布
Tips:如果要用该代码定义的每个场所的温度分布,是需要有假设的。因为温度分布随着时间的变化是会存在变化的。所以大家做的时候可以将时间考虑进去,根据时间求每个时刻的温度分布等
-
还有一个就是对病毒传播指数的定义,在该方案中我们是认为基本再生数就是我们的病毒传播指数。对label的定义不一样,很可能会影响到整道题的解法。
-
总之,比较片面,考虑得不是很全,有什么不懂的在底下留言就好,仅供学习交流(不一定正确)
Task4
等更新就好


