
- 1MAC下Burpsuite设置图标启动_burpsuite ico
- 2人格障碍诊断系统
- 3Win10系统Anaconda+TensorFlow+Keras 环境搭建教程_aconda拉取keras的环境
- 4微信小程序一次性订阅requestSubscribeMessage授权和操作详解
- 5URDF—ROS2_urtf ros2代码
- 6JAVA中几种常用JSON库性能比较_jmapper和structmap的性能比较
- 7(javaweb)请求响应postman
- 8Git-git diff命令结果解析_git diff结果
- 9【消息队列】RabbitMQ五种消息模式_rabbitmq 消息模式
- 10经典:创业的100个成功经验方法谈_成功之路分享一百个惊人变化
NCBI GEO王炸:GEO2R直接分析RNA-seq数据,几家欢喜几家愁?
赞
踩
GEO2R是NCBI GEO团队针对上传到GEO的芯片数据开发的一款在线差异分析、可视化作图工具,是广大数据分析人员的福音。然而,一直以来GEO2R仅针对芯片数据,对于越来越多的测序数据,只能下载所上传的matrix矩阵,进行分析,若没有上传表达矩阵,或者基因组版本不合适的话,往往还得下载原始数据重新分析,耗时耗力。
NCBI GEO团队推出了一项“王炸”更新:GEO2R可以直接分析RNA-seq测序数据了。
小伙伴们:喜大泪奔(喜闻乐见、大快人心、普天同庆、奔走相告)!
同事和我:工作要丢了么?时代抛弃我,连声招呼都不打啊!

1,NCBI GEO为什么要给我们准备RNA-seq count数据?
A major barrier to fully exploiting and reanalyzing the massive volumes of public RNA-seq data archived by SRA is the cost and effort required to consistently process raw RNA-seq reads into concise formats that summarize the expression results. To help address this need, the NCBI SRA and GEO teams have built a pipeline that precomputes RNA-seq gene expression counts and delivers them as count matrices that may be incorporated into commonly used differential expression analysis and visualization software.
大白话就是:SRA服务器上存了大量数据,一般人要分析,得下载,得比对,费时费力,反正数据在他们NCBI服务器上,服务器闲着也是闲着,直接套个pipeline把RNA-seq基因表达count处理好,放出来供大家使用。不知道又要革了多少人的命。
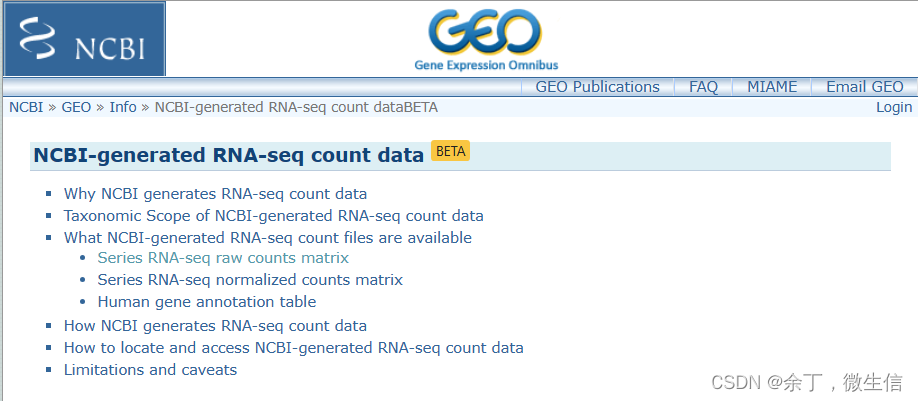
2,支持的物种
目前仅人类。小鼠的在路上,预计2023年秋。
3,数据类型:
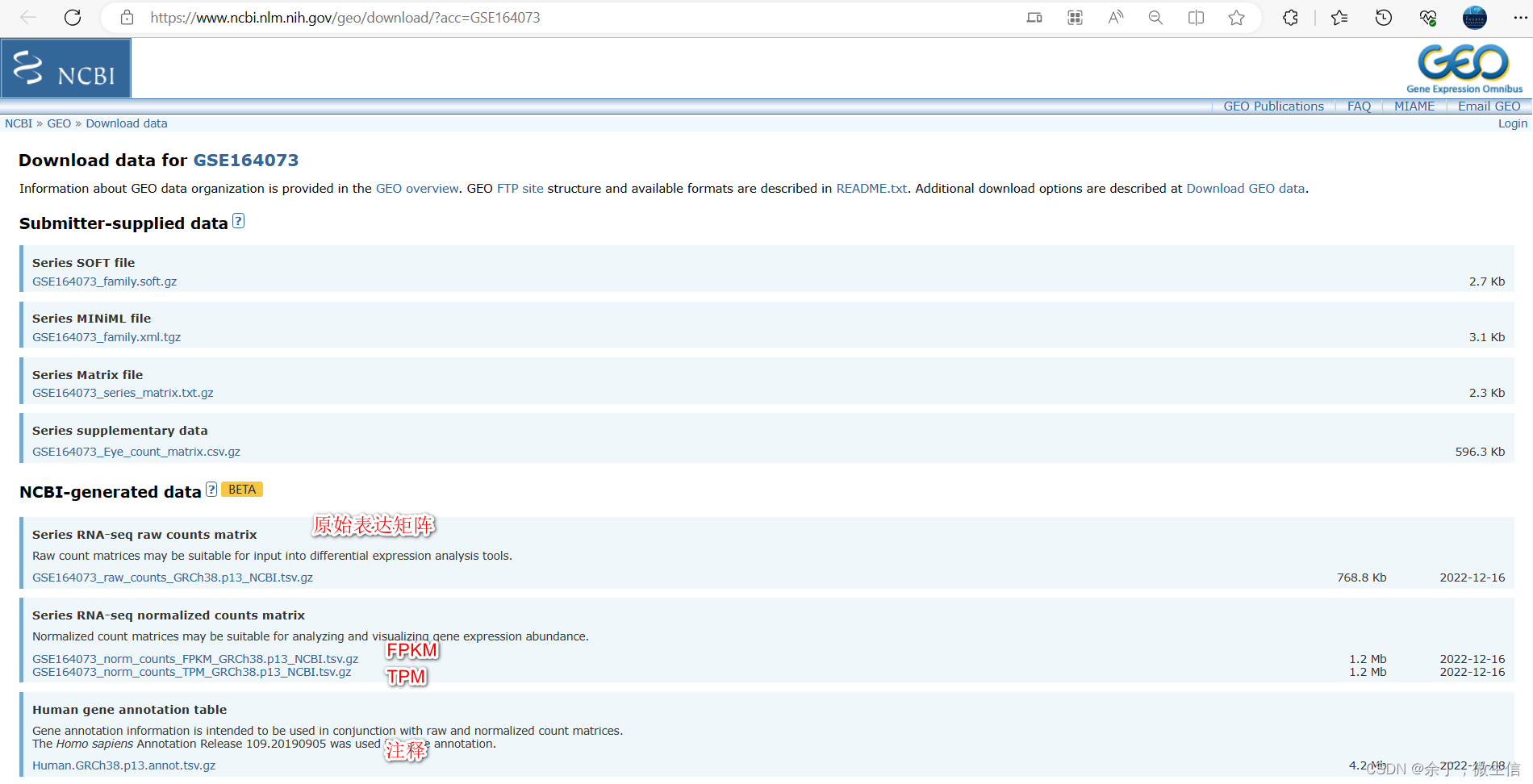
raw count:可以下载用DESeq2,edgeR或者limma voom进行后续的差异分析。
Normalized counts:根据测序深度和测序长度标准化的count,包括:FPKM (Fragments Per Kilobase Million,双端)、RPKM(Reads Per Kilobase Million,单端)和TPM(Transcripts Per Kilobase Million)
注释:基因id,gene symbol,descrption,gene ontology等。
4,分析流程
使用hisat2将物种为human,类型为transcriptomic的数据比对到GCA_000001405.15 参考基因组上。比对率大于50%的用featureCounts输出raw count文件。注释数据库用的Annotation Release 109.20190905.
5,如何下载?
https://www.ncbi.nlm.nih.gov/geo/download/?acc= GSE30970
直接换成自己的GSE号就行了
6,局限
GEO上的数据太杂了,GEO team没有对数据质量等深入检查(合不合适他们不管,先跑了再说),所以有以下局限性:
1)counts表达矩阵可能跟已发表文章的不一致
这个很容易理解,不同软件,甚至不同版本算出来的都不一样。
2)超过50%比对率的transcriptomic数据用于分析,所以质检可能很松,并且缺样品
上传的数据类型多样,可能不能直接比较,例如RNA-seq和RIP-seq都在矩阵里,但是不好直接比较。
3)Normalized矩阵文件并非充分标准化的。
对生信数据分析行业带来的冲击:
1)GEO的RNA-seq分析几乎要变得免费,无门槛了
2)有了表达矩阵,直接省了下载、比对的时间,极大提高了工作效率
3)有了统一标准?
4)大规模利用GEO数据的时代又来了
5)伦理问题
微生信助力高分文章,用户167000, 谷歌学术3100

