
热门标签
热门文章
- 1论文阅读:Language Models are Few-Shot Learners(巨无霸OpenAI GPT3 2020)_gpt-3.5: language models are few-shot learners
- 2Mongodb入门--头歌实验MongoDB 复制集 & 分片_头歌mongodb 复制集 & 分片
- 3关于程序员这个职业缺点总结_程序员述职缺点和不足
- 4【NLP屠夫系列】- NER之实战BILSTM_ner实战
- 5Linux系统中卸载Anaconda_linux卸载anaconda
- 6网络爬虫:Requests库入门
- 7Elasticsearch报错received plaintext traffic on an encrypted channel, closing connection Netty4TcpChann
- 8React如何使用 Ant Design(简单使用)_react antdesign
- 9Arduino 合宙 ESP32 S3 + OV2640 实现低成本SD存储卡相机(ESP32连接SD模块引脚)_esp 32 sd卡如何改引脚
- 10c++ ---------命令行获得和转换文件CommandLineToArgvW和GetCommandLineW()的使用
当前位置: article > 正文
diamond软件_BLAST本地比对太慢?不怕用diamond
作者:小丑西瓜9 | 2024-04-19 21:33:13
赞
踩
diamond blastx
在宏基因组学研究中,又或者你准备做大规模的系统进化的相关研究等等,需要分析了数百万个序列的reads,以确定来自环境的微生物样品的功能或分类学含量,找出对应reads同源的物种。这里重要的步骤是确定存在哪些基因,通常通过将翻译的DNA序列与蛋白质序列的参考数据库比对,例如NCBI非冗余(NCBI-nr)蛋白质数据库。长期以来,BLASTX因其高灵敏度而被认为是此标准的黄金标准工具。但是,BLASTX对于处理大批量的数据来说还是太慢了(就算你将你的蛋白质切成很小份并行分析,也需要一段长时间的运行,并且会耗费大量的计算机资源)。
今天的推文给大家介绍一下钻石(DIAMOND),目前的引用量已经超过1000,一款非常适合在处理大数据量的蛋白质或者核苷酸序列分析中替换BLASTX/BLASTP。据分析,当针对NCBI-nr数据库进行显着比对,预期值低于10 -3时,DIAMOND比BLAST比对大约快20,000倍于,并具两个工具有相似的灵敏度水平。
软件基本介绍
DIAMOND是一种高通量比对程序,可将DNA测序reads文件与蛋白质参考序列文件(如NCBI-nr)进行比较。它以C ++实现,旨在在多核服务器上快速运行。
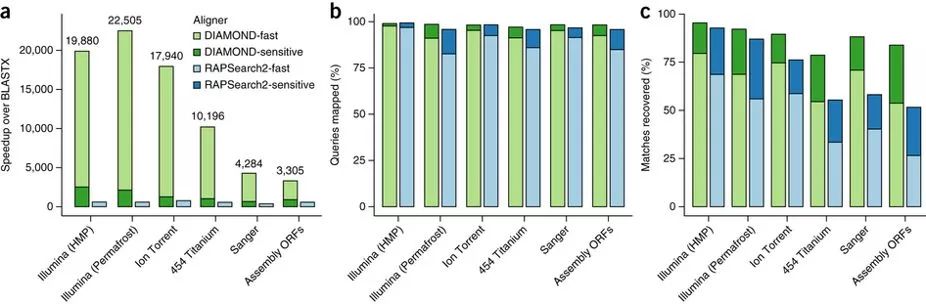
该程序明确地设计为,利用具有大内存容量和许多内核的现代计算机体系结构。那么为什么它那么快呢&#
声明:本文内容由网友自发贡献,不代表【wpsshop博客】立场,版权归原作者所有,本站不承担相应法律责任。如您发现有侵权的内容,请联系我们。转载请注明出处:https://www.wpsshop.cn/w/小丑西瓜9/article/detail/453750
推荐阅读
相关标签

