
- 1关于jsonobject类型的Null值怎么判断的问题,JSONObject$Null_jsonobject判断null
- 2INTELLIJ IDEA 项目(初始化/删除) GIT_idea删除git信息
- 3逻辑思维训练1200题-蓝桥杯计算思维参考_蓝桥杯计算思维u8
- 4优化算法|遗传算法求解作业车间调度问题(Python)_python实现遗传算法车间调度求解的代码
- 5mysql8 Incorrect datetime value: '0000-00-00 00:00:00'_mysql-connector-java-8.0.22 incorrect datetime val
- 6几道数据结构选择题_数据结构多选题
- 7唯一索引 oracle 空值,pgsql 组合唯一约束或唯一索引 null失效的情况
- 8【米哈游】2024秋招接近尾声,还没投递的抓紧上车啦!_深圳欣锐科技 计算机硕士 知乎
- 9FPGA实现的高效CIC滤波器:内插滤波器与数字信号多采样率处理_高效 数字滤波
- 10解决Win10找不到msvcp140_atomic_wait.dl文件_msvcp140-atomic-wait.dll
iMeta | 李香真/姚敏杰开发微生物共现网络比较流程
赞
踩
点击蓝字 关注我们
微生物共现网络比较分析指引

iMeta主页:http://www.imeta.science
研究论文
● 原文链接DOI: https://doi.org/10.1002/imt2.71
● 2023年1月10日,福建农林大学姚敏杰和中国科学院成都生物研究所李香真团队在 iMeta 在线发表了题为“A guide for comparing microbial co-occurrence networks ”的文章。
● 本研究针对当前微生物生态学的研究中多个共现网络难以比较分析的现状,创建了一个新的R语言包meconetcomp,并结合R microeco包的多种方法进行共现网络的比较分析。
● 第一作者:刘驰
● 通讯作者:李香真 (lixz@cib.ac.cn)、姚敏杰(yaomj@fafu.edu.cn)
● 合作作者:李超男、蒋艳琼、曾建雄
● 主要单位:福建农林大学资源与环境学院、中国科学院成都生物研究所
亮 点
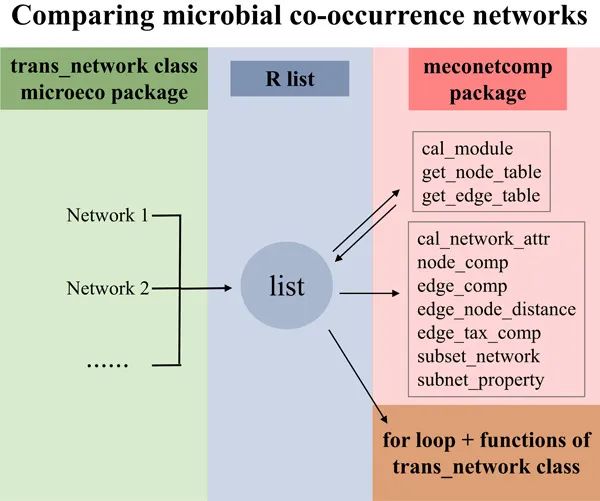
● 创建R meconetcomp 包用来方便地进行网络的比较分析
● 联合使用R microeco 包的多个类有助于网络比较
● 所提供的流程具有较高的灵活性和可拓展性
摘 要
本研究针对当前微生物生态学的研究中多个共现网络难以比较分析的现状,创建了一个新的R语言包meconetcomp,并结合R microeco包的多种方法进行共现网络的比较分析。
视频解读
Bilibili:https://www.bilibili.com/video/BV1M14y1u7Qf/
Youtube:https://youtu.be/8xDODhF37r8
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
微生物几乎无处不在,在各种环境下发挥着重要的功能,比如生物地球化学循环和宿主健康。自然环境下的微生物群落包括了大量的物种和数目。在微生物生态学的研究中,确定这些物种成员和他们的数目(丰度)是基础的任务。随着高通量测序的快速发展以及生物信息学软件的发展(如QIIME 2)使得这项工作逐渐变得容易。在获得OTU/ASV/species等的丰度后,后续分析可以使用相对成熟的R语言及相关的包进行操作。这些后续的分析在微生物生态领域与动植物生态领域中具有一定的相似性。但是微生物由于其个体极小、未培养的物种(基本属于未知)多、极高的多样性等特点使得很多生态学假设并不能很好地进行验证, 特别是跟微生物功能特征和相互作用相关的。在实际的数据分析工作中,通常需要运用大量的统计学方法来获取某个结论。R语言及相关方法的使用成为数据分析中的一个限制性的步骤。基于此所开发的R microeco包(https://github.com/ChiLiubio/microeco) 通过模块化的设计和多种方法的移植使得扩增子测序的后续数据分析变得更加容易。
在微生物群落的研究中,共现网络分析(co-occurrence network)常用来研究复杂微生物聚集下的微生物间的联系。这种方法是通过基于丰度数据来推测物种间的关联性(相关性等)。这与动植物生态学中所常使用的基于观测得到的物种间的相互作用关系所构造的网络有很大差别。测序的数据具有一些如结构性(序列数)、稀疏性(大量的0值)和推导物种间的直接关系比较困难等多个特点, 进而会影响网络的构建。基于相关系数的网络或许是最早用于微生物生态学各个领域(如土壤的研究)中的网络方法了。之后一些方法也陆续地发展出来,用于解决测序数据中使用相关性的方法所存在的非直接关联的问题,如SparCC、CCREPE和CCLasso;还有基于图论模型的方法来推断直接的关联,如SPIEC-EASI和FlashWeave等。另外BEEM-static的方法尝试将generalized Lotka-Volterra 模型拓展到横截面类型的数据中来推测物种间的互作关系。
伴随着生态学领域中网络方法的发展,一些研究者们开始担心网络方法在物种相互作用关系上的滥用问题。产生类似问题的一个主要原因是物种相互作用在群落生态的研究中具有极其重要的意义且难以研究。还有一个原因是不管使用的哪种网络构建方法,可能多多少少都会有一些互作关系能够包含在这种网络连接中。当然不管方法怎么改进,由于数据自身问题等所导致的一些互作关系是难以被方法所推测出来的,比如高阶的互作关系等。综合这些问题,基于物种间关联的共现网络在实际应用中不能被解释为物种间的互作关系。即使需要将这两者结合起来,也应该尽可能的基于一些生物学背景等来解释有多高的成分或者可能性可以将一些连接认为是互作关系所导致的。在动植物生态领域中,有研究也发现基于数据推测的关联性很难跟真实的物种互作关系所对应上。当前所常用的网络分析本质上还是经典的”checkerboard distribution”的另一种形式,也就是确定性过程的另一种呈现方式。在不同的体系中,生态系统的各种能导致群落变化的复杂性因素都会影响最终的网络表现形式,比如多重的非生物因素以及它们与生物学因素的交互。还有一个重要的点是不同的方法甚至是同一类型的网络构建方法,由于所依赖的统计模型等方法有差别,导致的结果也有差别。这给数据的解释也产生了一些困扰。
尽管网络方法已大量用于微生物生态学的研究中,单从生态学假设和解释网络指标背后的机制而言,还远远不够。通常网络的方法使用起来像黑盒子,缺少灵活性来进行网络分析方法与其它方法结合起来进行更多信息的挖掘。更加高效地使用网络方法离不开对网络构建方法和分析方法的流程和细节的深入了解。在实践中,通过结合多种网络构建方法来改善网络结构信息也具有一定价值。另外,对于复杂的实验设计和研究,网络的构建可能需要分别在不同的处理或者组别中进行,这也对于网络的比较提出了更高的要求。目前有一些在线工具(如MetagenoNets和iNAP)和R语言包(如igraph、NetCoMi和ggClusterNet)能够用来进行网络构建、分析和可视化。但网络的比较仍然是个难题。在创作这个指导手册的同时,我们也创建了R包meconetcomp(https://github.com/ChiLiubio/meconetcomp) 用于网络比较(Fig.1a),这项指导手册主要涉及microeco包 trans_network 类的使用,以及尽可能地结合其它方法来用于多个网络的批量分析及比较。
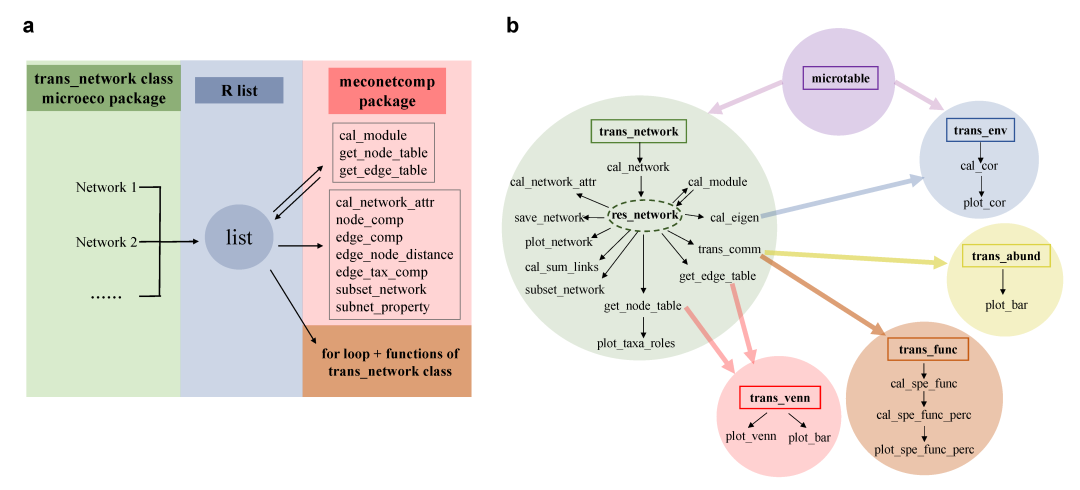
图1. 整体的框架图
a: 整个流程的分析策略图;b: 涉及到microeco包的方法示范图
方 法
定义
一个共现网络定义为边加权的网络图 G = (V, E),其中 V (node) 代表 特征(ASV/OTU/species), E (edge)代表网络中推测的连接。E 的权重(weight)代表了连接的强度。边的符号 (+ or -) 代表推测的正的或负的关联。本文出现的网络均为无向的网络。
依赖
R和R包的安装
如果R语言软件没有安装,使用者应首先下载并安装R (https://cran.r-project.org/)。同时强烈推荐安装Rstudio (https://posit.co/download/rstudio-desktop/) 来高效地使用R。
下面首先安装 R 包 meconetcomp (发音为 [miːkəunetˈkʌmp]) (Fig.1a)。整体的方法和函数等展现在流程图中(Fig.1b)。
# 安装 meconetcomp package (>= v0.2.0)
install.packages("meconetcomp")
然后安装几个CRAN上面的依赖包。
# 可视化使用
install.packages("networkD3")
install.packages("ggraph")
install.packages("rgexf")
# heatmap使用
install.packages("pheatmap")
# microeco包的依赖包之一
install.packages("aplot")
#用来进行 Duncan's new multiple range test
install.packages("agricolae")
准备数据集
两个已经内置在meconetcomp包里的数据集用来进行演示。一个是中国湿地土壤的16S rRNA gene的扩增子测序数据集。共有48个地点的200个样本。另一个是从R包ExperimentHub筛选出的宏基因组测序的分类谱数据集。共有198个具有喝酒习惯人群的粪便样本和92个物种。使用宏基因组数据有助于在物种水平上探索共现网络。
library(microeco)
library(meconetcomp)
# 使用magrittr包中的管道符
library(magrittr)
library(igraph)
library(ggplot2)
# 载入土壤16S扩增子测序数据集
data(soil_amp)
# 载入人粪便宏基因组分类谱数据集
data(stool_met)
▲ Trouble shooting:
• 用户如何导入自己的数据? 请查看教程中的详细说明 (https://chiliubio.github.io/microeco_tutorial/basic-class.html#microtable-class).
例子
不同组样本的网络构建示例
微生物生态的研究中,对不同组样本分别构建网络是一个常见的情景。这里使用soil_amp数据集作为示例来演示不同组样本的操作。相关性网络的构建通过调用在构建trans_network对象的时候计算的相关性及p值(储存在对象中的res_cor_p列表中)来实现。首先对不同的样本组分别创建trans_network对象。然后构造网络并将这些网络的对象储存于列表list中(Fig.1a)。数据集中的样本分成了3个组,包括内陆湿地(IW)、海岸带湿地(CW)和青藏高原湿地(TW)。
# 创建一个空的list来储存后面的网络对象
soil_amp_network <- list()
# 筛选IW组样本
# 使用 clone函数来完全复制一个soil_amp数据集 (R6 object)
tmp <-clone(soil_amp)
# 通过操作样本信息表来筛选样本
tmp$sample_table %<>%subset(Group == "IW")
# 将所有文件修整齐
tmp$tidy_dataset()
# 构建trans_network对象,根据需要过滤低丰度OTU
tmp <- trans_network$new(dataset = tmp, cor_method ="spearman", filter_thres =0.0003)
# COR_p_thres 代表p值阈值
# COR_cut 代表相关系数阈值
tmp$cal_network(COR_p_thres =0.01, COR_cut =0.6)
# 将网络对象放置于列表中
soil_amp_network$IW <- tmp
# 重复上面的操作,筛选TW组样本并建网络对象
tmp <-clone(soil_amp)
tmp$sample_table %<>%subset(Group == "TW")
tmp$tidy_dataset()
tmp <- trans_network$new(dataset = tmp, cor_method ="spearman", filter_thres =0.0003)
tmp$cal_network(COR_p_thres =0.01, COR_cut =0.6)
soil_amp_network$TW <- tmp
# 重复上面的操作,筛选CW组样本并建网络对象
tmp <- clone(soil_amp)
tmp$sample_table %<>%subset(Group == "CW")
tmp$tidy_dataset()
tmp <- trans_network$new(dataset = tmp, cor_method ="spearman", filter_thres = 0.0003)
tmp$cal_network(COR_p_thres =0.01, COR_cut = 0.6)
soil_amp_network$CW <- tmp
▲ Trouble shooting:
• 对于较大数据集的分析, 使用”use_WGCNA_pearson_spearman = TRUE” 能够加速计算。
不同网络构建方法的示例
上面的操作也可以用于不同的网络构建方法来进行方法学上的研究。下面的示例使用stool_met数据集来完成。除了这些示例中使用的网络构建方法,还有其它方法可以选择。对于相关网络中的其它方法(如SparCC)请查看trans_network$new中的cor_method参数。对于其它网络构建方法(比如SpiecEasi, FlashWeave和BEEM-static)请查看cal_network函数中的network_method参数。
# 同样的先创建一个空的list
stool_met_network <-list()
使用Bray-Curtis index (1 - dissimilarity) 来做一个网络。
stool_met_bray <- trans_network$new(dataset = stool_met, cor_method = "bray")
stool_met_bray$cal_network(COR_cut =0.4)
stool_met_network$stool_met_bray <- stool_met_bray
# 删除非必要的中间变量来节省内存空间
rm(stool_met_bray)
然后基于Pearson 和 Spearman 相关方法的不同参数来构建不同的网络作为例子。
# Pearson (R >= 0.4, P < 0.05)
stool_met_pear_r4 <- trans_network$new(dataset = stool_met, cor_method ="pearson")
stool_met_pear_r4$cal_network(COR_p_thres =0.05, COR_cut =0.4)
stool_met_network$stool_met_pear_r4 <- stool_met_pear_r4
rm(stool_met_pear_r4)
# Pearson (R >= 0.6, P < 0.05)
stool_met_pear_r6 <- trans_network$new(dataset = stool_met, cor_method ="pearson")
stool_met_pear_r6$cal_network(COR_p_thres =0.05, COR_cut =0.6)
stool_met_network$stool_met_pear_r6 <- stool_met_pear_r6
rm(stool_met_pear_r6)
# Spearman (R >= 0.4, P < 0.05)
stool_met_spear_r4 <- trans_network$new(dataset = stool_met, cor_method ="spearman")
stool_met_spear_r4$cal_network(COR_p_thres = 0.05, COR_cut =0.4)
stool_met_network$stool_met_spear_r4 <- stool_met_spear_r4
rm(stool_met_spear_r4)
# Spearman (R >= 0.5, P < 0.05)
stool_met_spear_r5 <- trans_network$new(dataset = stool_met, cor_method ="spearman")
stool_met_spear_r5$cal_network(COR_p_thres =0.05, COR_cut =0.5)
stool_met_network$stool_met_spear_r5 <- stool_met_spear_r5
rm(stool_met_spear_r5)
# Spearman (R >= 0.6, P < 0.05)
stool_met_spear_r6 <- trans_network$new(dataset = stool_met, cor_method ="spearman")
stool_met_spear_r6$cal_network(COR_p_thres = 0.05, COR_cut =0.6)
stool_met_network$stool_met_spear_r6 <- stool_met_spear_r6
rm(stool_met_spear_r6)
▲ Trouble shooting:
• 查看帮助文档,请使用命令: help(trans_network)。
网络模块化
网络模块化是对紧密连接的节点进行分类。在trans_network的cal_module函数中,有许多基于igraph包的方法可用来计算模块。meconetcomp包提供了相同名称的单独的 cal_module 函数来批量进行模块化的计算。
# 使用默认的igraph的 cluster_fast_greedy的方法
soil_amp_network %<>%cal_module(undirected_method ="cluster_fast_greedy")
stool_met_network %<>%cal_module(undirected_method ="cluster_fast_greedy")
网络的拓扑学属性
R包 meconetcomp提供的cal_network_attr函数可以同时计算列表中各个网络的整体的拓扑学属性然后返回一个汇总的表格。在计算完后,列表中的每个trans_network对象中都分别储存了一个 res_network_attr 表格。
tmp <- cal_network_attr(soil_amp_network)
# 保存下结果
write.csv(tmp, "soil_amp_network_attrubutes.csv")
tmp <-cal_network_attr(stool_met_network)
write.csv(tmp, "stool_met_network_attrubutes.csv")
节点和边的属性
包microeco中trans_network类的get_node_table和get_edge_table分别用来计算或提取节点和边的属性信息。R meconetcomp包提供了相似的函数名来批量操作列表中的所有网络。在计算完后,列表内部每个网络对象中都会有 res_node_table 和 res_edge_table 表格。后续的一些分析会用到这些基础信息。
soil_amp_network %<>% get_node_table(node_roles = TRUE) %>% get_edge_table
stool_met_network %<>%get_node_table(node_roles =TRUE) %>% get_edge_table
网络的可视化
这里所提供的网络可视化部分是为了对单个的网络进行图形查看,而非用来进行比较分析。在R中的可视化有静态的和动态的方式。其中静态的方法适合网络较小的情况。
# 缺省方法是 'igraph'
stool_met_network[["stool_met_spear_r5"]]$plot_network(method ="igraph")
stool_met_network[["stool_met_spear_r5"]]$plot_network(method ="igraph", vertex.size =4, edge.arrow.size = .5, vertex.label.dist =1, vertex.label.color ="gray50")
stool_met_network[["stool_met_spear_r5"]]$plot_network(method ="ggraph", node_color ="Phylum")
# 保存ggraph的图片到电脑
tmp <-"stool_met_network_plot_ggraph"
dir.create(tmp)
for(i in names(stool_met_network)){
# use the default colors
g1 <- stool_met_network[[i]]$plot_network(method ="ggraph", node_color ="Phylum")
ggsave(paste0(tmp, "/", i, ".pdf"), g1, width =9, height =7)
}
动态网络的方法基于 networkD3 包,特别适合快速查看较大的网络。
stool_met_network[["stool_met_spear_r5"]]$plot_network(method ="networkD3", node_color ="Phylum")
另一类可视化方式是保存文件然后使用 Gephi 软件 (https://gephi.org/)。
# 保存每个网络到文件夹
tmp <-"stool_met_network_gexf"
dir.create(tmp)
for(i in names(stool_met_network)){
stool_met_network[[i]]$save_network(filepath =paste0(tmp, "/", i, ".gexf"))
}
比较不同网络的节点
比较网络中的节点是一个重要的内容。这里的操作策略是,使用meconetcomp包中的node_comp函数将所有网络中的节点提取出来并转化成一个新的microtable对象。然后使用trans_venn类来查看节点的重叠性 (Fig.1b)。
# 搜索和合并网络对象中的 res_node_table
tmp <- node_comp(soil_amp_network, property = "name")
# 使用trans_venn类来计算节点的交集
tmp1 <- trans_venn$new(tmp, ratio = "numratio")
g1 <- tmp1$plot_venn(fill_color = FALSE)
ggsave("soil_amp_node_overlap.pdf", g1, width = 7, height = 6)
# 使用常用的 jaccard distance 来量化网络间的差别
tmp$cal_betadiv(method = "jaccard")
tmp$beta_diversity$jaccard
# stool_met 数据集因为网络数目较多,使用柱状图可视化
tmp <- node_comp(stool_met_network)
tmp1 <- trans_venn$new(tmp)
# 这里移除没有交集的部分来可视化
tmp1$data_summary %<>% .[.[, 1] != 0, ]
g1 <- tmp1$plot_bar()
ggsave("stool_met_node_overlap.pdf", g1, width = 10, height = 6)
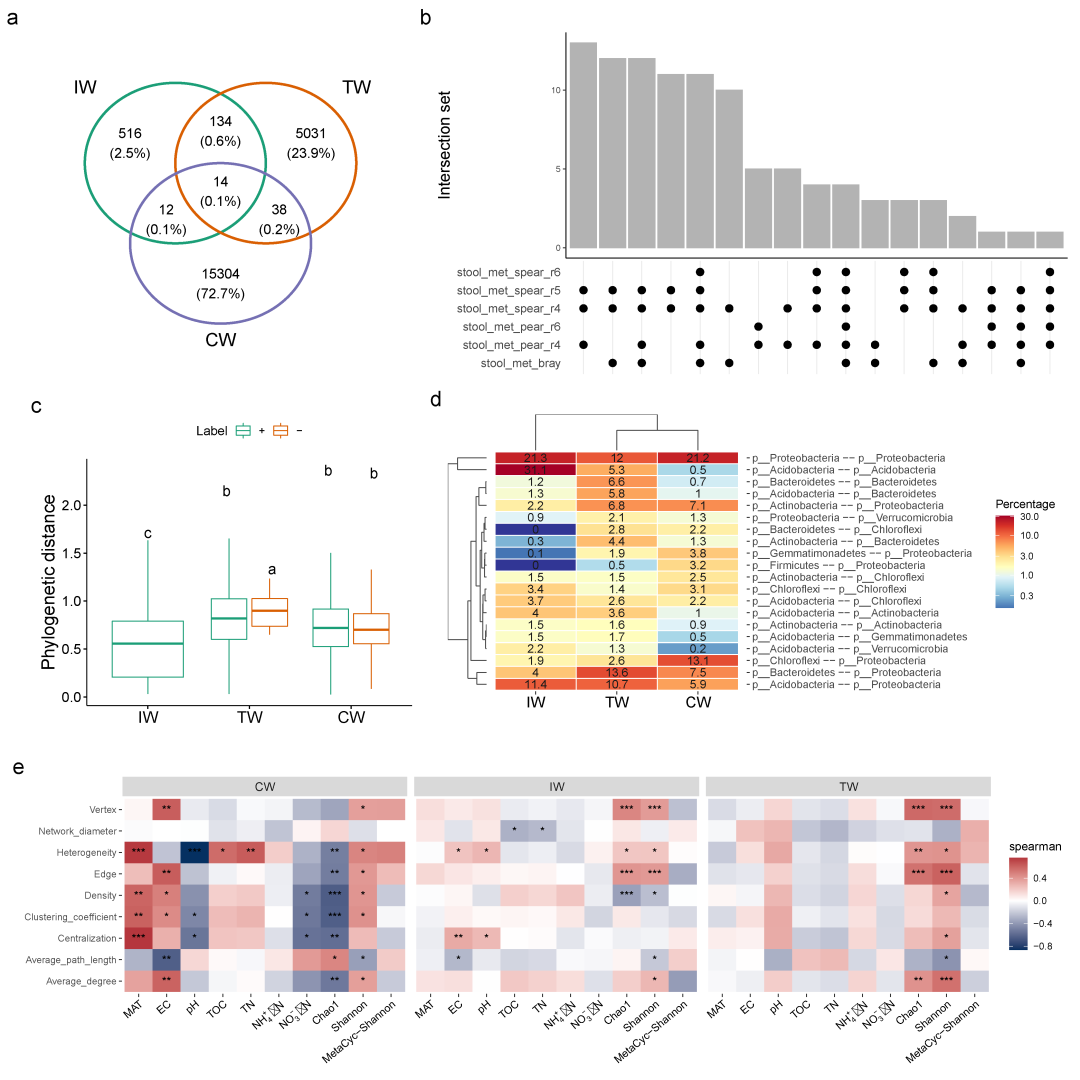
图2. 流程中部分结果展示
a: 土壤数据集所做3个网络的边的交集的数目;b: 人粪便数据集中不同网络方法得到的边的交集数目;c: 土壤数据集所做各个网络的节点间(边连接的)系统发生距离的分布情况;d: 正相关的边连接的节点的分类信息在门水平的展示(正相关的与总的边的数目比);e: 土壤样本的子网络拓扑学属性与理化和多样性之间的相关性。
比较不同网络的边
比较不同网络的边的操作逻辑与上面的节点的比较相似。所不同的是其所需使用的是meconetcomp包的edge_comp函数。
# 产生图 Fig.2a
# 搜索和合并网络对象中的 res_edge_table
tmp <- edge_comp(soil_amp_network)
# 通过trans_venn产生交集
tmp1 <- trans_venn$new(tmp, ratio = "numratio")
g1 <- tmp1$plot_venn(fill_color = FALSE)
ggsave("soil_amp_edge_overlap.pdf", g1, width = 7, height = 6)
# 使用jaccard distance量化网络间在边上的差别
tmp$cal_betadiv(method = "jaccard")
tmp$beta_diversity$jaccard
# 图Fig.2b
tmp <- edge_comp(stool_met_network)
tmp1 <- trans_venn$new(tmp)
tmp1$data_summary %<>% .[.[, 1] != 0, ]
# 只显示共有的
tmp1$data_summary %<>% .[grepl("&", rownames(.)), ]
g1 <- tmp1$plot_bar()
ggsave("stool_met_edge_overlap.pdf", g1, width = 10, height = 6)
提取不同网络重叠的边
提取不同网络所共有的边并将其通过meconetcomp包的subset_network函数将其转化为一个新的trans_network对象。
# 搜索和合并网络对象中的 res_edge_table
tmp <- edge_comp(soil_amp_network)
tmp1 <- trans_venn$new(tmp)
# 转换集合信息到新的 microtable 对象
tmp2 <- tmp1$trans_comm()
# 使用subset_network提取结果,这里提取三个网络的交集
# 使用 colnames(tmp2$otu_table) 来查找名字
Intersec_all <-subset_network(soil_amp_network, venn = tmp2, name ="IW&TW&CW")
# Intersec_all 是一个新的 trans_network 对象
# 举例:保存Intersec_all为gexf格式并使用gephi查看
Intersec_all$save_network("Intersec_all.gexf")
比较节点间的系统发生距离
物种间的系统发生距离与许多生态学问题具有密切的联系。在网络中,一条边所连接的节点可能来自于亲缘关系较近的或者较远的分枝。一个网络所有的边所具有的系统发生关系的距离的分布情况可能跟数据以及网络构建的方法有关系。在meconetcomp包中创建了一个R6类edge_node_distance来进行距离的转换和差异分析 (Fig.2c)。参数dis_matrix的输入内容必须是具有行名和列名(网络节点名一致的名字,方法会对名字自动筛选排序)的对称性的矩阵。因此这个方法也可以用来比较其它距离,比如物种间的生态位重叠性(Levin’s niche overlap)等。
# soil_amp中总的OTU数目太多,在前面低丰度的OTU并没有用于网络构建
# 这里去掉这些低丰度的,以免计算太慢
# 直接取出每个网络对象中的丰度数据中的名字即可
node_names <- unique(unlist(lapply(soil_amp_network, function(x){colnames(x$data_abund)})))
# 复制数据集,然后过滤
filter_soil_amp <- clone(soil_amp)
filter_soil_amp$otu_table <- filter_soil_amp$otu_table[node_names, ]
filter_soil_amp$tidy_dataset()
# 获得系统发生距离矩阵
phylogenetic_distance_soil <- as.matrix(cophenetic(filter_soil_amp$phylo_tree))
# 调用edge_node_distance类
tmp <- edge_node_distance$new(network_list = soil_amp_network, dis_matrix = phylogenetic_distance_soil, label = c("+", "-"))
# 差异检验
tmp$cal_diff(method = "anova")
# Fig.2c
g1 <- tmp$plot(boxplot_add = "none", add_sig = TRUE, add_sig_text_size = 5) + ylab("Phylogenetic distance")
ggsave("soil_amp_phylo_distance.pdf", g1, width = 7, height = 6)
# stool_met 数据集演示
phylogenetic_distance_stool <- as.matrix(cophenetic(stool_met$phylo_tree))
tmp <- edge_node_distance$new(network_list = stool_met_network, dis_matrix = phylogenetic_distance_stool, label = c("+", "-"))
tmp$cal_diff(method = "anova")
g1 <- tmp$plot(boxplot_add = "none", add_sig = TRUE, add_sig_text_size = 5, xtext_angle = 30) + ylab("Phylogenetic distance")
ggsave("stool_met_phylo_distance.pdf", g1, width = 7, height = 6)
# 这里分模块进行距离的转换来作为另一个例子
tmp <- edge_node_distance$new(network_list = soil_amp_network, dis_matrix = phylogenetic_distance_soil,
label = "+", with_module = TRUE, module_thres = 10)
tmp$cal_diff(method = "anova")
g1 <- tmp$plot(boxplot_add = "none", add_sig = TRUE, add_sig_text_size = 5) + ylab("Phylogenetic distance")
ggsave("soil_amp_phylo_distance_modules.pdf", g1, width = 8, height = 6)
节点的拓扑学属性
基于within-module connectivity (Zi) 和 among-module connectivity (Pi)对节点进行划分和可视化。分为 Module hubs (模块内部具有高度的连接性, Zi > 2.5 & Pi ≤ 0.62); Connectors (节点主要连接不同模块, Zi ≤ 2.5 & Pi > 0.62); Network hubs (分别具有Module hubs和Connectors的特征, Zi > 2.5 & Pi > 0.62); Peripherals (节点有稀少的连接而且基本是在模块内部, Zi ≤ 2.5 & Pi < 0.62)。
tmp <- "stool_met_node_roles"
dir.create(tmp)
for(i in names(stool_met_network)){
stool_met_network[[i]]$res_node_table %<>% .[!is.na(.$taxa_roles), ]
g1 <- stool_met_network[[i]]$plot_taxa_roles(add_label = TRUE, add_label_group = c("Network hubs", "Module hubs"), label_text_size = 4, label_text_italic = TRUE)
ggsave(paste0(tmp, "/", i, ".pdf"), g1, width =7, height =5)
}
比较不同网络的节点分类学属性
与上面系统发生距离相似的是,研究节点的分类学信息同样具有重要意义。使用meconetcomp包中的edge_tax_comp函数能够获得在不同的分类学水平上每个边所连接的节点的分类信息。这对于研究网络特征和节点的生物学意义非常有帮助。
# 在门水平上进行示例
soil_amp_network_edgetax <- edge_tax_comp(soil_amp_network, taxrank = "Phylum", label = "+", rel =TRUE)
# 过滤比例较低的类别
soil_amp_network_edgetax <- soil_amp_network_edgetax[apply(soil_amp_network_edgetax, 1, mean) > 0.01, ]
# Fig.2d
g1 <- pheatmap::pheatmap(soil_amp_network_edgetax, display_numbers = TRUE)
ggsave("soil_amp_edge_tax_comp.pdf", g1, width = 7, height =7)
stool_met_network_edgetax <-edge_tax_comp(stool_met_network, taxrank ="Phylum", label ="+", rel = TRUE)
g1 <- pheatmap::pheatmap(stool_met_network_edgetax, display_numbers =TRUE)
ggsave("stool_met_edge_tax_comp.pdf", g1, width =7, height =7)
模块的eigengene分析
为了评估网络模块与非生物因素间的关系,使用trans_network中的cal_eigen函数进行模块的eigengene分析并使用相关分析来研究其与环境因子的相关性。这里使用soil_amp数据集内置的环境因子信息来完成。
# 分别分析每个网络
tmp <- "soil_amp_module_eigen"
dir.create(tmp)
for(i in names(soil_amp_network)){
# module eigengene 分析
soil_amp_network[[i]]$cal_eigen()
# 筛选样本,因为是分组构建的网络
tmp1 <- clone(soil_amp)
tmp1$sample_table %<>% base::subset(Group == i)
tmp1$tidy_dataset()
# 创建 trans_env 对象来进行相关分析,选择一些环境因子
tmp2 <- trans_env$new(dataset = tmp1, env_cols =8:15)
tmp2$cal_cor(add_abund_table = soil_amp_network[[i]]$res_eigen)
g1 <- tmp2$plot_cor()
ggsave(paste0(tmp, "/", i, ".pdf"), g1, width = 7, height =5)
}
不同模块的物种组成和功能特征组成
类trans_network中的trans_comm函数能够将节点转化为一个新的microtable对象,进而可以分析其它内容。这里使用soil_amp数据集来展示模块的组成以及使用FAPROTAX数据库(主要是生物地球化学循环相关的功能)来预测OTU的功能性状。
tmp_dir <- "soil_amp_module_composition"
dir.create(tmp_dir)
for(i in names(soil_amp_network)){
# 为了进行完整演示,重新获取下 res_node_table
soil_amp_network[[i]]$get_node_table()
# trans_comm 转换节点信息表
tmp <- soil_amp_network[[i]]$trans_comm(use_col ="module", abundance =FALSE)
# 转换成二元信息
tmp$otu_table[tmp$otu_table >0] <- 1
# 过滤一些物种数目太少的模块
tmp1 <- soil_amp_network[[i]]$res_node_table$module %>% table %>% .[. >= 10] %>% names
tmp$sample_table <- tmp$sample_table[rownames(tmp$sample_table) %in% tmp1, ]
tmp$tidy_dataset()
tmp$cal_abund()
# 使用trans_abund来进行可视化
tmp2 <- trans_abund$new(tmp, taxrank = "Phylum", ntaxa = 10)
tmp2$data_abund$Sample %<>%factor(., levels =rownames(tmp$sample_table))
# 显示比例信息
g1 <- tmp2$plot_bar(xtext_type_hor =TRUE, color_values = RColorBrewer::brewer.pal(12, "Paired")) +ylab("OTUs ratio (%)")
ggsave(paste0(tmp_dir, "/taxa_", i, ".pdf"), g1, width =7, height = 5)
# 功能预测
tmp3 <- trans_func$new(tmp)
tmp3$cal_spe_func(prok_database ="FAPROTAX")
# 获取百分比数据
tmp3$cal_spe_func_perc(abundance_weighted =FALSE)
# 作图
g1 <- tmp3$plot_spe_func_perc(order_x = tmp$sample_names())
ggsave(paste0(tmp_dir, "/trait_", i, ".pdf"), g1, width =9, height = 6)
}
比较网络子集的拓扑学属性
在这一部分,使用soil_amp数据集来根据每个样本中存在的OTU来提取网络子集并计算网络拓扑学属性。操作使用meconetcomp包的 subnet_property 函数来完成。
# 根据样本中的feature名提取子网络并计算拓扑学属性
tmp <- subnet_property(soil_amp_network)
# 整理数据集
# 使用第二列的样本名作为行名
rownames(tmp) <- tmp[, 2]
# 删除其它信息
tmp <-tmp[, -c(1:2)]
# 载入整理好的土壤理化表格和多样性信息
data(soil_measure_diversity)
tmp1 <- trans_env$new(dataset = soil_amp, add_data = soil_measure_diversity)
tmp1$cal_cor(use_data ="other", by_group ="Group", add_abund_table = tmp, cor_method ="spearman")
# 产生 Fig.2e
g1 <- tmp1$plot_cor()
ggsave("soil_amp_subnet_property.pdf", g1, width =11, height = 5)
讨 论
网络比较流程具有一定的灵活性和可拓展性
在这个流程中,我们主要介绍了一系列的网络比较分析方法。这些操作有别于其它(如NetCoMi)网络分析和比较的工具。这个流程充分使用了R list、microeco包和新建的meconetcomp包的优点。最大的优势就是这里所能比较的网络数目并没有限制。用户还可以根据其它需要对所转换的结果进行其它比较分析。当前并没有网络构建方法标准可供使用。之前有研究发现相关性方法的灵敏性和精确性均会随着数据集而变化。这里也发现了相似的情况(Fig.2b以及其它使用stool_met_network 数据集产生的结果)。所以我们认为除了一些经验性的方法选择之外,用户使用这里的流程对网络方法进行比较分析,进而选择能够更好解释数据背后的生态学和生物学问题的方法是可行的。
将网络结果与生态学规律和生物学背景相联系
将环境因素对于网络结构的影响成分解析出来是一个非常吸引人的主题。然而由于标准数据集的缺失,类似的操作可能会在可行性上打折扣。一项最近的研究发现,在土壤中很少有正相互作用关系是由于微生物间的代谢互营所导致的。所以在更加异质的环境下,共现网络的解释应该向生态学规律所靠拢。在这些情况下,通过结合其它不同的生态统计学方法来揭示网络所蕴含的属性是非常重要且可行的。例如,soil_amp数据集所呈现的不同网络的系统发生距离的分布情况 (Fig.2c) 跟这些不同区域的群落所受到的选择性(可能造成谱系聚集等)有很大关系。而且,从不同的非生物的因素可能会影响这些群落子网络的拓扑学属性 (Fig.2e)中也能够看出这一点。另外,针对异质性不太高的样本(这里人粪便样本均为饮酒人群的),使用多个网络构建方法来获取鲁棒性更高的边 (Fig.2b)进而解释边代表的生物学意义(比如互作关系)则是可行的(流程里用的多个方法仅是为了方便展示从而简单调整的相关性的参数,实际情况下可能需要考虑其它网络构建方法,见文中说明)。
全面展示网络分析和比较可能超出了本研究的范围。这个流程中的例子想通过结合R中的list和microeco包以及基于microeco包所创建的meconetcomp包展示可行的网络比较和高效率的分析。为了使得流程聚焦于网络分析,这项研究并没有考虑太多数据整理相关的操作,比如重抽样、稀疏数据过滤和网络构建参数等。这些操作可能在用户的分析中具有一定的重要性。
代码和数据可用性
除了 meconetcomp 包中的数据外, 这项研究流程所涉及的数据也储存在了GitHub (https://github.com/ChiLiubio/network_protocol) 和 Gitee (https://gitee.com/chiliubio/network_protocol) 中。
引文格式:
Liu, Chi, Chaonan Li, Yanqiong Jiang, Raymond J. Zeng, Minjie Yao, and Xiangzhen Li. 2022. “A guide for comparing microbial co‐occurrence networks.” iMeta. e71.
https://doi.org/10.1002/imt2.71
作者简介

刘驰(第一作者)
● 福建农林大学农业资源与环境专业博士生
● 研究方向为农业微生物组,相关学术成果已发表于iMeta、Geoderma、FEMS Microbiology Ecology等期刊。其中高引论文一篇。以主要作者之一开发了microeco、file2meco和meconetcomp等R语言包

姚敏杰(通讯作者)
● 福建农林大学教授,博导
● 研究方向为土壤微生物和微生物生态学,迄今发表论文40余篇,其中以第一/通讯作者在Soil Biology & Biochemistry、Geoderma、FEMS Microbiology Ecology、Catena、Applied Microbiology and Biotechnology、生态学报和微生物学报等期刊发表论文20篇。主持国家自然科学基金项目3项、国家重点研发任务1项、国家自然科学基金区域联合项目课题1项、四川省重点研发项目1项、横向项目10余项。福建省引进高层次人才(C类),福建省土壤肥料学会理事

李香真(通讯作者)
● 中国科学院成都生物研究所研究员,博导
● 主要从事废弃物生物处理技术、土壤微生物、环境基因组学方面的研究工作。建立了环境基因组和生物信息学平台,建设了土壤微生物组数据库(http://egcloud.cib.cn/)。2021、2022年入选全球前2%顶尖科学家榜单。主持国家科技部973项目课题、支撑项目子课题、国家自然科学基金项目、中国科学院项目、企业合作项目等20余项。在Environmental Sciences & Technology、Soil Biology and Biochemistry、Water Research、Nature communications、Global Ecology and Biogeorgraphy等刊物上发表论文180余篇。曾经及现在担任Annals of Microbiology副主编, Land Degradation and Development、Soil Ecology Letters等期刊的编委。
更多推荐
(▼ 点击跳转)
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法
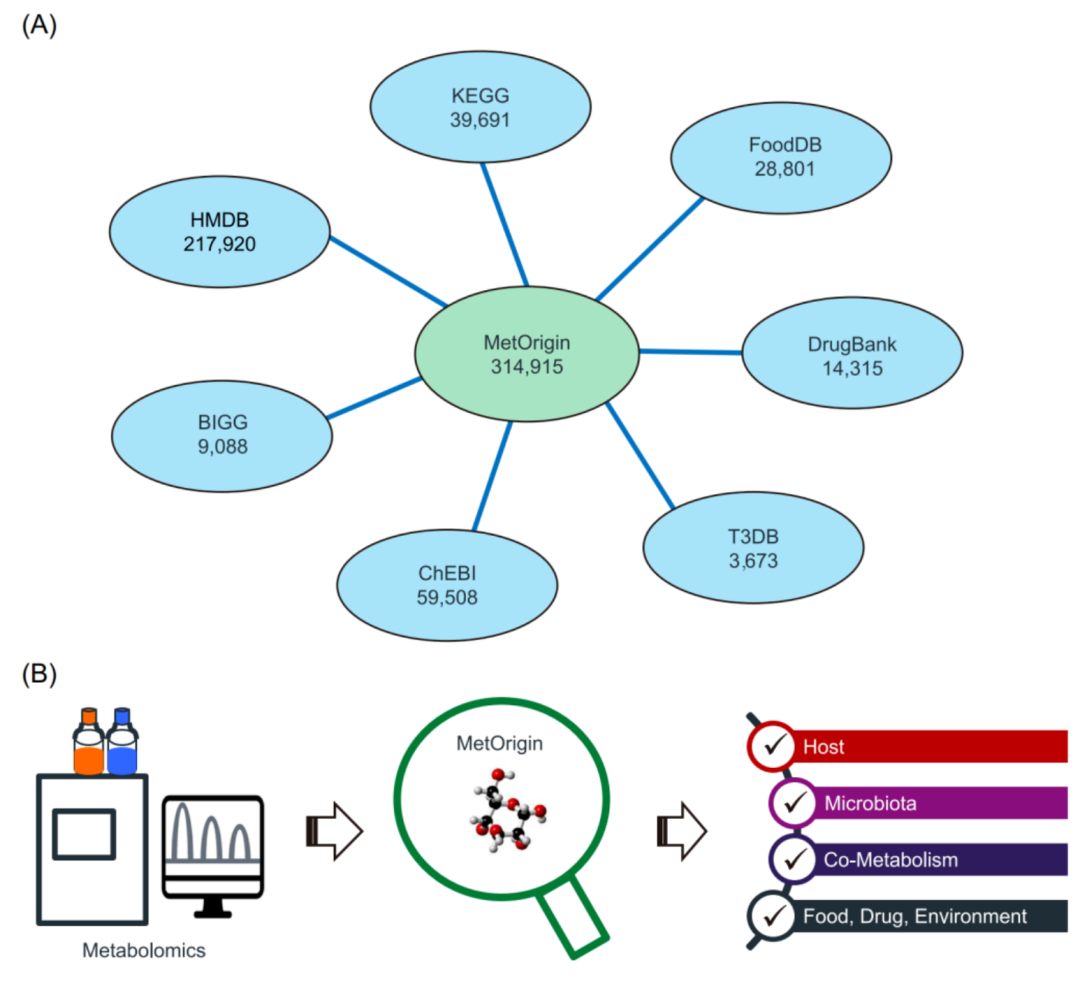
iMeta | 浙大倪艳组MetOrigin实现代谢物溯源和肠道微生物组与代谢组整合分析

第1卷第1期

第1卷第2期

第1卷第3期

第1卷第4期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science

