
- 13.26总结
- 2你真的会使用logcat吗_can't create wucai.log: read-only file system logc
- 3uni-app开发---4.首页
- 4Java中创建线程的多种方式实例
- 5Android中kernel内核模块编译执行_安卓kernel编译
- 6flutter-无法运行到模拟器_flutter run 运行模拟器报错 could not resolve all files fo
- 7Python123 期末题库_2.表达式求值(2)成绩5开启时间2023年09月28日 星期四 10:00折扣0.8折扣时间
- 8AI基础知识扫盲
- 9神经网络:正则化 范数 Droupout BN 方差 偏差 距离 损失 梯度 指标 学习率 超参数 归一化 标准化 激活 过拟合抑制 数据增强 标签平滑_dropout 批标准化 神经网络
- 10CentOS升级版本及内核_centos最新版本
VESTA官方手册 | 第六章:设置单相参数_vasp原子位移
赞
踩
第六章 设置单相参数
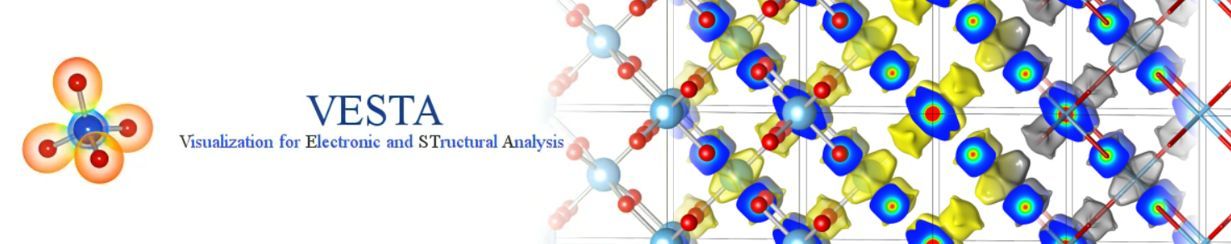
选择File菜单中的New Structure…,创建新结构。选择Edit菜单Edit Data | Phase…,编辑已有数据。在这两种情况下将分别出现名为New Data或Edit Data的内容一致的对话框(图6.1)。此对话框包含五个选项页面:
· Phase相
· Unit cell单位晶胞
· Structure parameters结构参数
· Volumetric data体数据
· Crystal shape晶体外形
在对话框顶部,显示所选相的序号和标题。
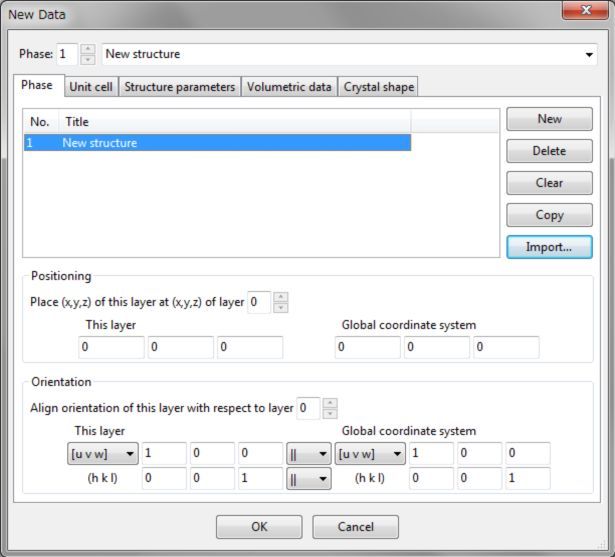
图6.1:New Data对话框
6.1 相的定义
在New Data对话框的Phase选项卡(图6.1)中,可以添加、删除、复制或导入体相数据,并在同一图形区叠加显示。选择列表中的一行并单击Title列可以编辑体相的标题。当处理单一相时,除了编辑标题外此选项卡无其他作用。在叠加两个或两个以上的相时,可使用Positioning和Orientation框。在同一图形区域内可视化多个相数据的方法可参见第7章。
6.2 对称性和单位晶胞
Edit Data对话框的Unit cell选项卡可设置晶格常数和结构的对称性(图6.2)。
图6.2:Edit Data对话框的Unit cell选项卡
6.2.1 晶体对称性和空间群
选择晶系
System列表用于过滤Space group列表中的空间群符号列表。当选择此列表框中的项目时,只会在Space group列表框中突出显示属于所选晶系的空间群。在System列表框中列出了以下10项。
Molecule分子
Custom自定义
Triclinic三斜
Monoclinic单斜
Orthorhombic正交
Tetragonal四方
Trigonal三方
Hexagonal六方
Cubic立方
Rhombohedral菱面体
如果选择Molecule,将采用直角坐标系而非分数坐标系,导致不再存在单位晶胞。因此,空间群和晶格参数设置的文本框和列表框将不能使用。当选择Custom时,无法指定空间群设置,但可以手动定义对称操作(见第36页)。
选择空间群
空间群编号和标准符号列在Space Group列表框中。列表中只突出显示选定晶系的空间群,但可以选择任何空间群。在选择空间组群,将更新System和Setting的列表框将被更新。
设置空间群
所选空间群的可设置的参数列于Setting列表框中。列表框中可用的设置基于《国际晶体学表International Tables for Crystallography》A卷中的给定。
对于一些空间群,Setting在列表框中包含更多设置。在三斜晶系中,可选择复式格子A、B、C、I、F、R,设置编号分别为2、3、4、5、6、7(表6.1)。
表6.1:两种三斜空间群中的非标准化设置
在单斜晶系中,特征轴a、b和c的设置均可作为内置设置(表6.2)。
表6.2:单斜空间群中的设置编号
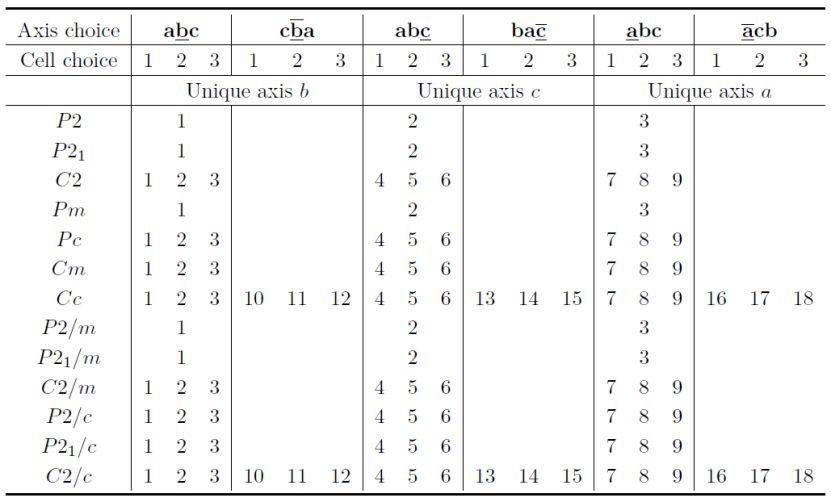
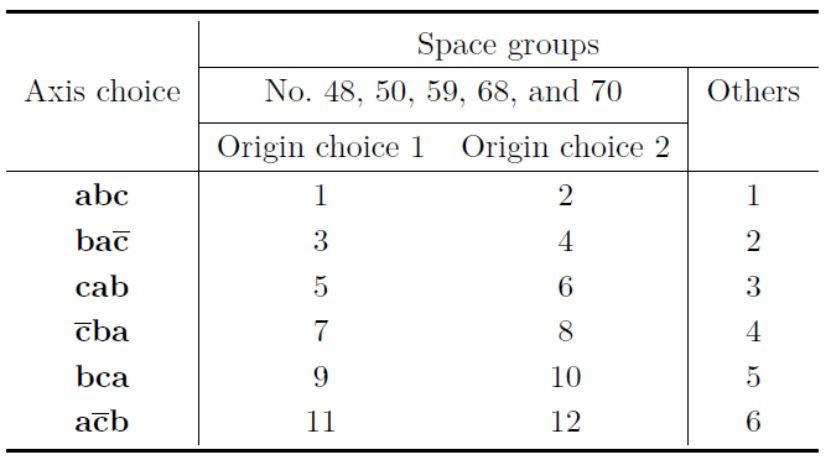
在正交晶系中,可选择设置以下六种之一,即abc、ba-c、cab、-cba、bca和a-cb,设置编号分别为1、2、3、4、5和6。在正交空间群中,有两种原点选择,分别使用奇数和偶数设置编号选择原点1和2。并且,设置编号1和2是abc的轴选择,设置编号3和4是ba-c的,等(表6.3)。
表6.3:正交空间群的晶轴选择
当晶轴变化时,如三方晶系中的菱面体或六方的晶轴,晶格参数自动转换。
在更改设置编号时,需选择是否需要保留结构参数或三维几何结构(见下一部分)。
6.2.2 改变空间群设置时的响应
Setting下的列表框能够设置改变空间群设置时的响应。有两个选项:
更新结构参数保持3D几何构型
保持结构参数不变
如果使用第一个选项(默认),当改变空间群的设置数或指定变换矩阵时,将转换晶格参数和分数坐标,从而保持晶体的结构几何构型。当使用第二个选项时,晶格参数和分数坐标保持不变,而改变几何构型。
如果在图形区域中显示出正确的结构,但相对于当前的空间群设置,另一个空间群更加合适,则选择更新结构参数来保持三维几何构型,改变空间群设置。第二个选项通常用于在读取了CIF等文件后,当正确的设置编号未被VESTA识别时,更改一个设置编号。在这种情况下,晶格参数和分数坐标是正确的,而对称操作是错误的。然后,选择保持结构参数不变并选择正确的设置编号。当从结构-数据文件中记录的数据不能唯一确定一个设置编号时,VESTA假设在两个原点可选的中心对称空间群中设置编号为2 (一个以原点为反演中心的设置),在其他所有空间群中设置编号为1。
注意,即使结构参数不变,在当前的空间群设置中,某些晶格参数也可能按照对其施加的约束而改变。
6.2.3 晶格参数
在Lattice parameters框中,晶格常数a、b、c以Å为单位,α、β和γ以°为单位。根据晶系自动约束用户输入的晶格参数。晶格参数的标准不确定度( s.u.)用来计算几何参数的s.u.。当Crystal system选定为Molecule时,对于基于笛卡尔坐标的结构数据,禁用此框中的文本框。
6.2.4 自定义对称操作
单击Customize…自定义对称操作。将出现带有编辑模式的Equivalent Positions对话框(图6.3),在此对话框中显示一系列一般等效位置。当列表中的一个等效位置被选中时,对应的对称操作以矩阵形式显示在对话框左上角。如果在其中一个文本框中输入一个值,则更新列表中相应的等效位置的表示。单击New按钮添加新的对称操作,在列表中选择一项并单击Delete按钮删除对称操作。单击Clear按钮将删除除标识操作以外的所有对称操作。
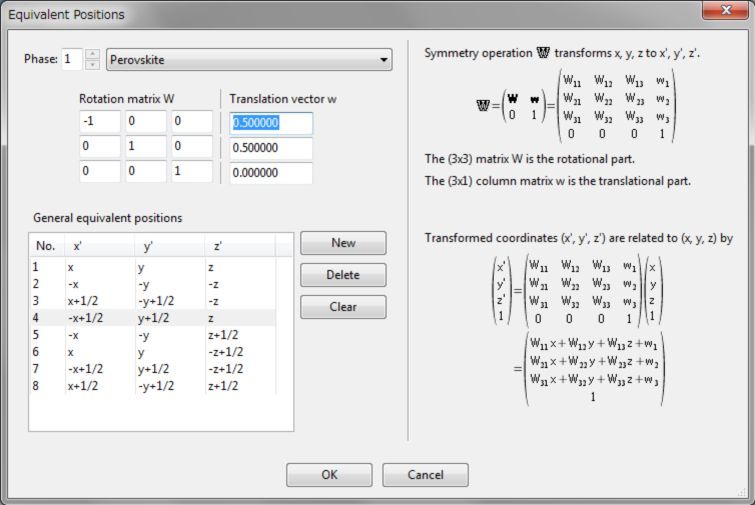
图6.3:带有编辑模式的Equivalent Positions对话框
6.2.5对称操作的约化
单击Remove symmetry按钮,即可通过将晶体的对称性降低到P1,产生所有原子均为虚拟独立位点的单位晶胞。当空间群变为对称性较高的空间群时,或者当单位晶胞变换为较小的单位晶胞时(见6.2.6),同一原子位置可能由两个或两个以上的位置通过对称变换产生。在这种情况下,可以通过单击Structure parameters选项卡中的Remove duplicate atoms按钮来删除冗余位置。
6.2.6 单位晶胞的变换
单击Transform...按钮,打开Unit Cell Transformation对话框(图6.4)。可使用此对话框,设置3×3旋转矩阵P和3×1平移向量p,从而转换晶轴。单击对话框中的View general positions按钮,打开Equivalent Positions对话框,查看转换后单位晶胞的一般等效位置和对称操作(见14.1)。单击Initialize current matrix按钮,将转换矩阵重置为单位矩阵。目前,这一操作无法取消。当勾选了“Normalize the fractional coordinates归一化分数坐标”选项时,转换后单位晶胞新的原子坐标将在0到1范围内归一化。
接下来,将对Unit Cell Transformation对话框进行较为详细的说明和用例分析。
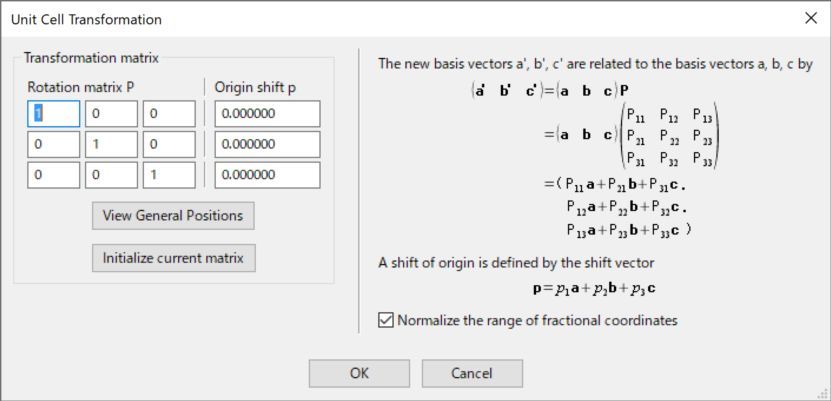
图6.4:Unit Cell Transformation对话框
非惯用空间群设置
如要将结构从惯用设置转换为非惯用设置,在打开Unit Cell Transformation对话框之前,需在Edit Data对话框Unit cell选项卡中,选择模式Update structure parameters to keep 3D geometry。在此模式下,晶格参数和分数坐标将会自动转换,从而保持当前晶体结构的几何构型不变。如果选择了Keep structure parameters unchanged模式,将仅有对称操作会转换为相应新设置,而晶格参数和分数坐标将保持不变。
制定原点位移p为转换后单位晶胞的新原点。例如如果设置了(0.25, 0.25, 0.25)为原点位移,惯用设置中的坐标(0.25, 0.25, 0.25),将在转换后的非惯用单位晶胞中成为(0, 0, 0)。
转换后晶轴的方向a'、b'、c'使用3×3矩阵P中的列矢量设置,即P的第一、二、三列,分别表示a'、b'、c'。例如,下面的转换矩阵可用于交换a和b轴。
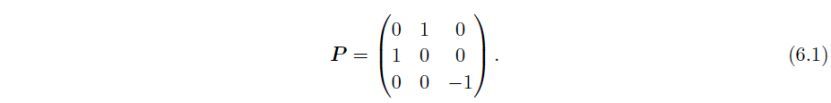
注意,在这种情况下,c轴的取向是倒置的,否则在非中心对称空间群中,变换后的结构将成为原结构的镜像。一般来说,当P的行列式det ( P )为负时,晶体坐标系由右手系向左手系转变,反之亦然。当P设置为这样的矩阵时,会出现一个警告对话框来确认是否进行下一步操作。
图6.5:结构手性转换的警告对话框
单位晶胞的体积V通过下式转换为V':
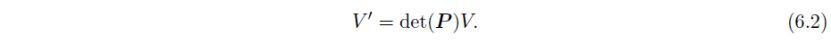
因此除非det(P)为1,否则转换后单位晶胞的体积将会变化。
整个变换包括一次旋转和一次原点平移,可以用一个4×4变换矩阵P表示,它由3×3的旋转矩阵P、3×1的平移向量p和行向量o = ( 0, 0, 0 ) 组成:
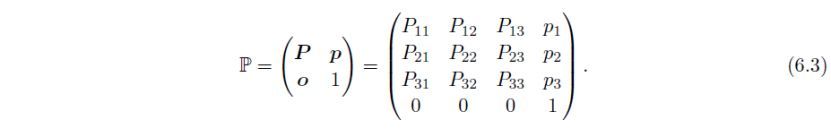
然后,下列各晶体学变量,即原始平移向量a,b,c,倒易晶格矢量a*,b*,c*,度量张量G,倒易晶格度量张量G*,原子坐标X,对称操作W和各向异性原子位移参数β,进行如下所述的变换。
原始平移向量a,b,c根据P进行转换:
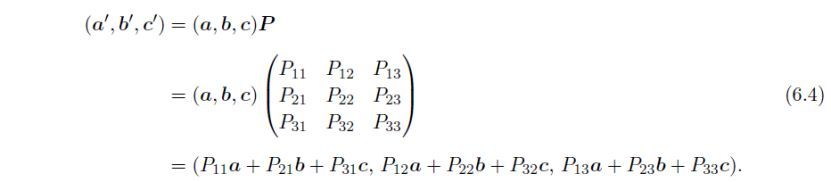
原点平移,即(0, 0, 0),使用平移向量t定义为:
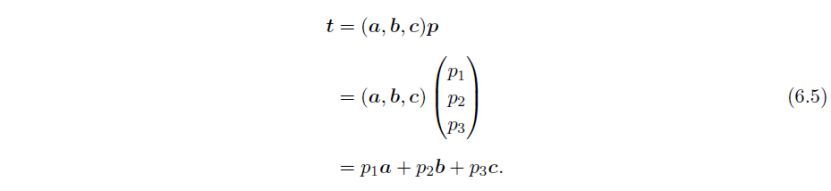
设A是在一组正交向量x、y、z的基础上,原始平移向量的表示矩阵a、b、c:
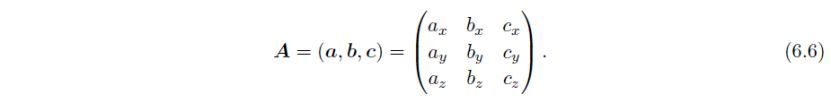
通过式(6.4)进行的转换可以写作:
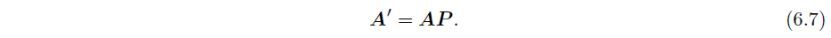
或者,由于G的唯一定义,使用度量张量G往往优于直接使用A。另一方面,由于正交基矢x,y和z可以任意选择,A的表示数是无限的。直接晶格的度量张量(矩阵)定义为:
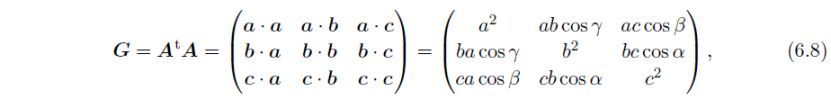
其中t表示转置。通过下式将G转换为G':
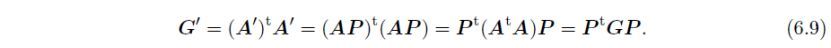
同样,倒易基矢a*,b*和c*的表示矩阵定义为:
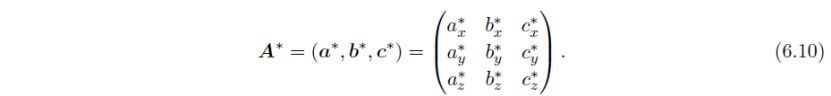
直接基矢和倒易基矢之间的关系为:
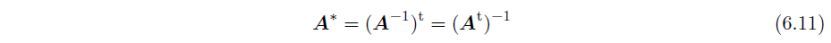
以及:
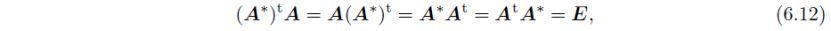
此处E为单位矩阵:
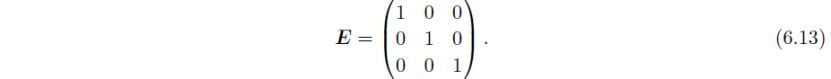
倒易晶格的度量张量定义为:
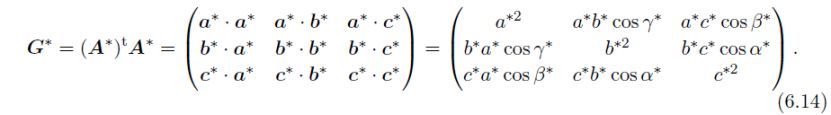
通过下式将G*转换为G*':
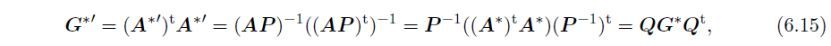
此处。
实空间中的x、y、z坐标利用4×4变换矩阵Q变换为:
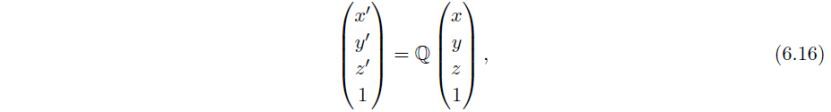
此处
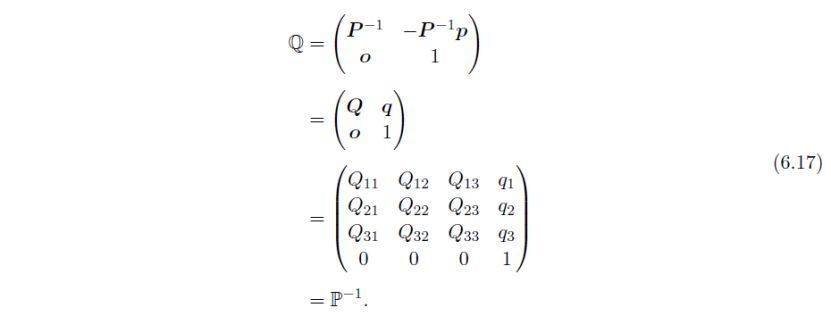
式(6.16)可简化表示为:
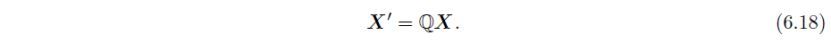
通过4×4对称操作矩阵W将坐标为x、y、z的点X,对称变换为坐标为和的等效点:
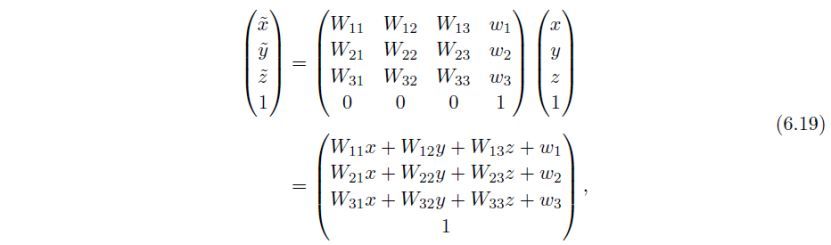
或简化表示为:
矩阵Q分别将X和转换为X'和':
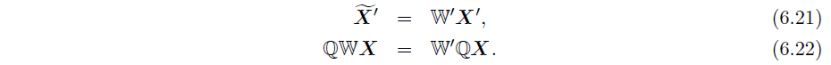
接下来,利用旋转矩阵转换对称操作W:
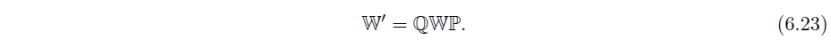
令为式(6.17)中定义的矩阵Q的转置矩阵。各向异性原子位移参数β,变换为:
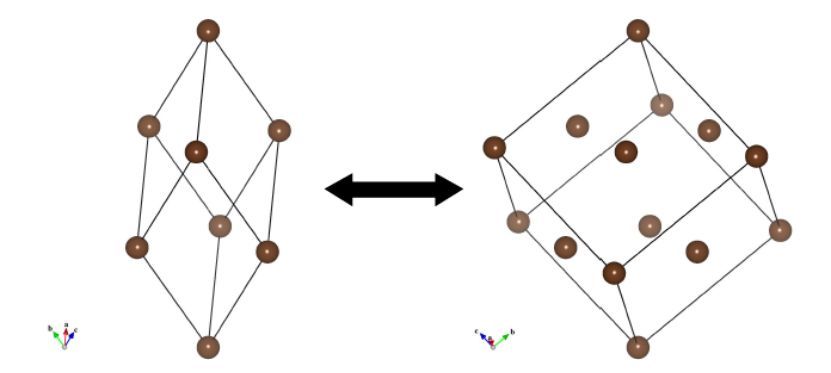
图6.6:简单格子和面心立方格子之间转换的实例
超结构和子结构的创建
转换矩阵也被用来转换(简单格子)⇄(复式格子)(图6.6),并创建超结构和平均子结构。要转换结构,必须在打开Unit Cell Transformation对话框之前,在Edit Data对话框的Unit cell选项卡中选择Update structure parameters to keep 3D geometry模式。
当P的行列式中,det ( P )大于1时,变换后的单位晶胞比原单位晶胞大,而当det ( P )小于1时,变换后的单位晶胞比原单位晶胞小。在这种情况下,会出现一个对话框来确认是否打算更改单位晶胞体积(图6.7)。
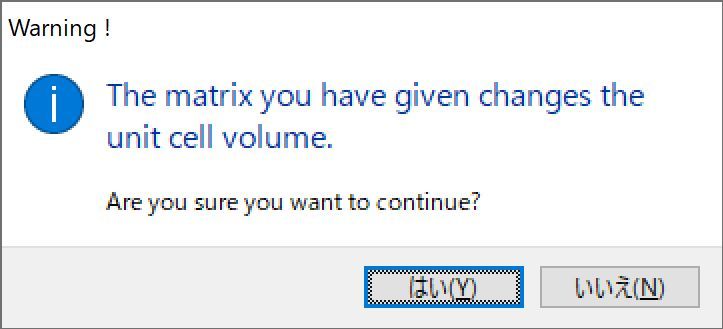
图6.7:单位晶胞转换确认对话框
当指定了一个det(P) > 1的旋转矩阵,或者虽然det(P) = 1,但转换后的晶胞包含其他晶格点,会显示对话框询问转换结构的方式(图6.8)。有以下三个选项:
Add new equivalent positions to a list of symmetry operations在对称操作列表中添加新的等效位置
Search atoms in the new unit cell and add them as new sites在新的单位晶胞中搜索原子并将其添加为新位点
Do nothing不进行操作
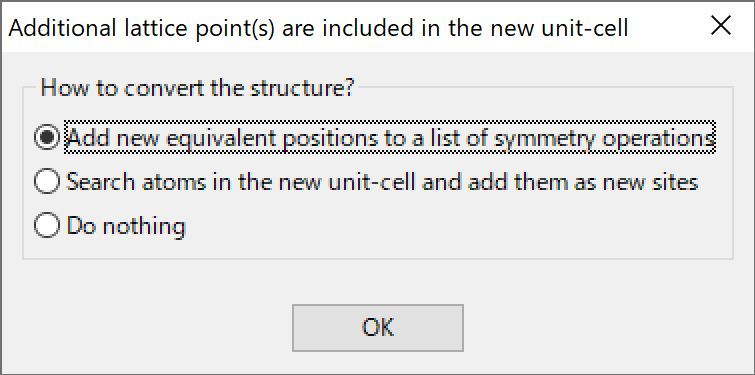
图6.8:对话框显示如何创建更大的单位晶胞的选项
如果选择了第一或第二项,将保持初始数据的结构几何构型不变。如果选择了第三项,在新的单位晶胞中的一些原子将消失。在选择第一项时,将通过检查下式,寻找附加的对称操作W'来创建超结构,W'具有(0, 0, 0)和(1, 1, 1)之间的平移分量:
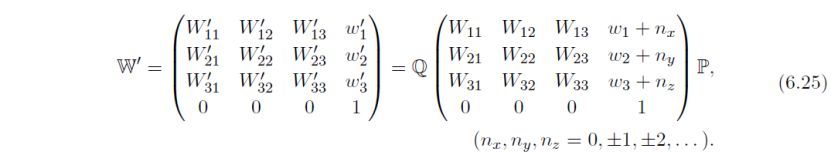
如果选择第二项,会通过检查下式,搜索在(0, 0, 0)和(1, 1, 1)之间存在的额外位点,创建超结构:
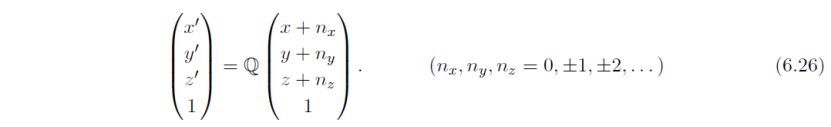
注意变换后的新单位晶胞应与当前使用的空间群对称性一致。否则,在创建超结构之前应单击Remove symmetry按钮,将对称性降低到P1,在超结构创建之后输入新的空间群。如果det ( P ) < 1,则同一位置可能来自两个或两个以上的位点。在这种情况下,可以通过单击Structure parameters选项卡中的Remove duplicate atoms按钮(见第48页)来删除冗余位点。
6.2.7 磁性结构
VESTA完全支持1651个具有磁性的空间群。勾选Magnetic structure复选框,可使用磁性空间群。然后,Setting列表框的标题将更改为BNS Setting,它显示了选定空间群的磁性空间群列表。磁性空间群的标记有两类:Belov–Neronova–Smirnova (BNS)标记和Opechowski–Guccione (OG)标记。VESTA使用BNS标记,因为BNS标记的对称操作的描述针对平移部分不超过1的磁单位晶胞。磁性结构的原子坐标、位移参数和占据率与常规结构完全相同(见6.3节)。原子的磁矩在Vectors对话框中设置(见第9.1节)。
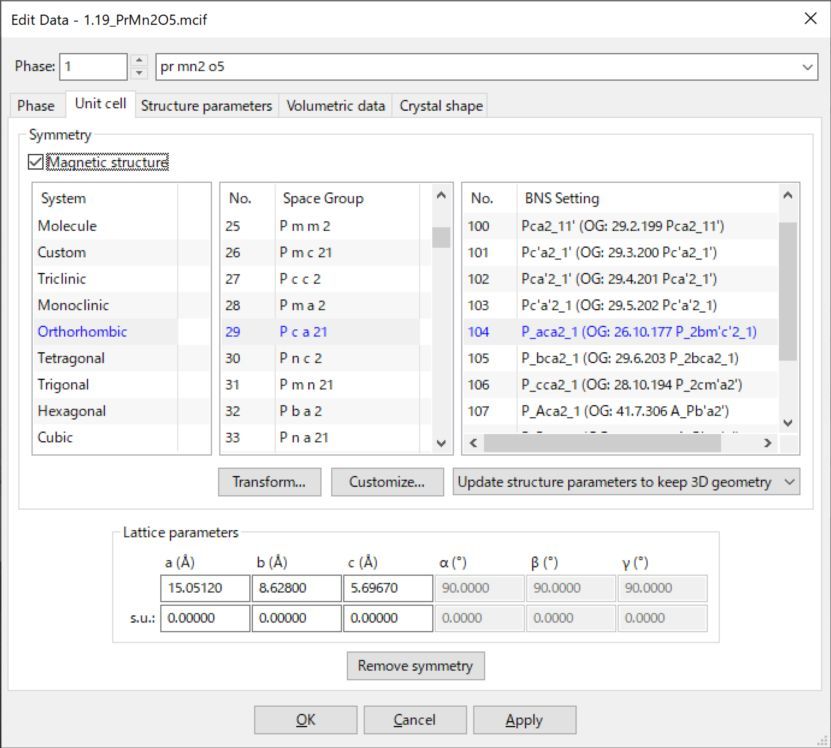
图6.9:Edit Data对话框的Unit cell页面,其中勾选了Magnetic structure选项
6.3 结构参数
Edit Data对话框的Structure parameters选项卡(图6.10)用于输入和编辑于原子结构有关的参数。可在该选项卡上半部分的文本框中,输入或编辑以下结构信息:
Symbol...:元素符号(最多两个字符)。
Label:位点名称(最多六个字符)。
Charge:形式电荷(氧化数;可选)。
x、y、z:分数坐标x、y、z(无量纲)。
s.u.(x)、s.u.(y)、s.u.(z):分数坐标的标准不确定度(默认值:0)。
Occ:占据率(无量纲;可选)。
B或U:各向同性原子位移参数,B或U(单位为Å2;可选)。
U11、U22、U33、U12、U13、U23:各向异性原子位移参数Uij(单位为Å2;可选)。
beta11、beta22、beta33、beta12、beta13、beta23:各向异性原子位移参数βij(无量纲;可选)。
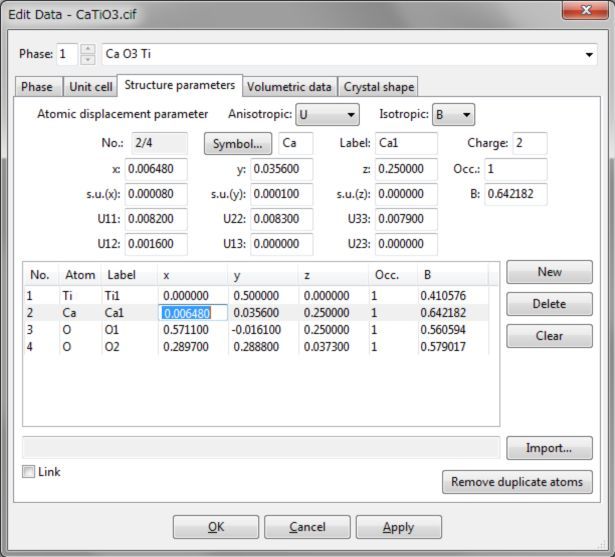
图6.10:Edit Data对话框的Structure parameters选项卡
非对称单位晶胞中的位点列表显示在页面的下半部分。当列表中没有选择位点时,文本框无法编辑。当列表中某项被选中时,文本框将更新,以显示与所选位点相关的数据。还支持列表项的实时编辑,如图6.10所示。
如要添加新的位点,首先单击New按钮,再在文本框中编辑文本。在光标位于另一个文本框或另一个GUI控件后,该文本框中的修改将立即应用到所选的位点。在文本框中需要输入数值的情况下,如果输入的文本不是有效的数值,或者文本框为空,将放弃此次修改。因此,如果意外更改了文本框中的一个值,可以通过在将光标移到另一个GUI控件之前删除文本框中的全部文本来恢复原始值。
选择列表中的一项,单击Delete按钮,或按下键盘上的Delete键,可以删除一个位点。单击Clear按钮,将删除列表中的所有位点。
6.3.1 元素符号和标签
对于每个位点,元素符号最多输入两个字符,标签最多输入9个字符。单击Symbol...按钮,可以从Periodic Table对话框(图6.11)中选择元素符号。
图6.11:Periodic Table对话框
6.3.2 形式电荷
严格来讲,形式电荷不属于结构参数。然而可使用形式电荷,根据键价参数估算键长(见11.4.3)、从键长估算电荷分布(见11.4.3)、用傅里叶方法计算静电位势和Madelung能量(见14.5)。
6.3.3 分数坐标
对于特殊位置,输入分数,如1/4、1/2、1/3,或应给出足够位数的有效数字,如0.333333、0.666667。
当使用RIETAN-FP精修的各向异性原子位移参数绘制位移椭球时,对于每个Wyckoff位置,需要输入对应于《国际晶体学表(第一卷)》中描述的第一个等效位置的分数坐标。例如,对于空间群P4/mmm (No. 123)中的4i位点,需输入(0, 1/2, z),而非(1/2, 0, z)。
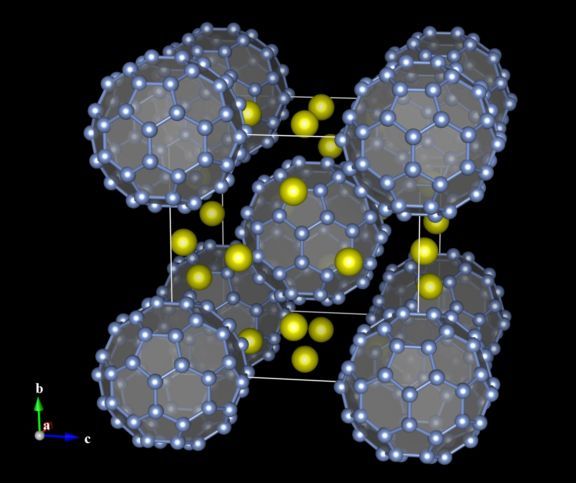
图6.12:Cs6C60的晶体结构,其中C60以半透明的多面体表示。在C60的中心添加了一个占据率为g = 0的虚拟位点X。通过在Bonds对话框中指定X-C键,创建了C60多面体。C-C键也通过Search molecules模式搜索,以实心圆柱体表示为多面体的边。创建化学键和多面体的方法详见第8章。
6.3.4 占据率
占据率的定义以位点完全占据的占据率为1个单位,位点为虚拟位点的占据率为0。如果一个位点的占据率小于1个单位,则占据在该处的原子显示为占据圆圈图。如果单色球体优先于双色球体占据,为了方便起见,将占据率改为1个单位。
如果一个位点的占据率为零,则该位点被视为一个虚拟位点,这个虚拟位点不被真实结构中的任何原子占据。虚拟位点和连接它们的键都不显示在屏幕上。虚拟位点可应用于辅助可视化显示,例如,笼状结构的多孔晶体可显示为固态多面体,而不会产生大量不必要的化学键(图6.12)。
6.3.5 原子位移参数
Debye–Waller因子Tj(h),常称为温度因子,包含在结构因子F(h)的表达式中,用以表示原子j的静态和动态位移的影响(见公式14.8和14.11)。原子的位移有两种不同的形式:各向异性原子位移和各向同性原子位移。
各向异性模型
各向异性Debye–Waller因子Tj(h),定义为:
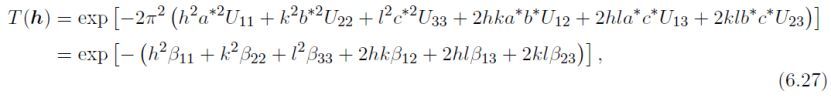
此处a*、b*、c*、α*、β*和γ*为倒易晶格参数。
各向异性原子位移参数的类型需在Anisotropic列表框中设置:“None”为省略各向异性原子位移参数,“U”输入Uij ( U11、U22、U33、U12、U13和U23 ),“beta”输入βij(β11、β22、β33、β12、β13和β23)。利用该列表框,可以将Uij转换为βij,反之亦然。
利用其它方式定义的各向异性原子位移参数必须利用上述定义转换为Uij或βij。
如使用RIETAN-FP的Rietveld方法对βij进行精修,每个位点的βij都必须满足对其施加的线性约束。
各向同性模型
各向同性Debye–Waller因子Tj(h),定义为:
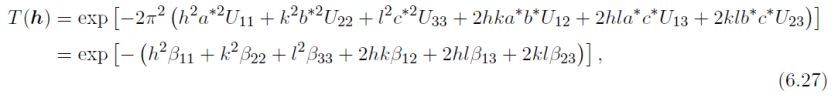
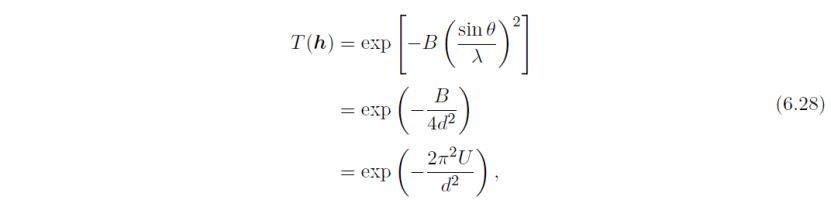
此处B和U为各向同性原子位移参数(U = B/8π2), θ为Bragg角,λ为X光或中子波长,d为晶面间距。B和U与均方位移<u2>有关,沿垂直于反射平面的方向:
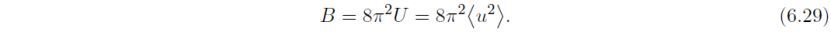
例如,如果B的值为3Å2,其对应的位移约为0.2Å。
各向同性原子位移参数的类型需在Isotropic列表框中设置。利用该列表框,可以将U转换为B,反之亦然。
利用其它方式定义的各向异性原子位移参数必须利用上述定义转换为U或B。
对于已输入各向异性原子位移参数Uij或βij的晶体学位点,可由Uij、a*、b*、c*,和度量张量G计算等效各向同性原子位移参数Beq或Ueq:
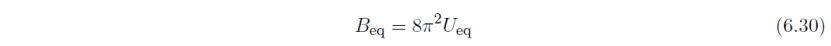
在重新打开Edit Data对话框后,在Structure parameters选项卡中,Beq和Ueq即被视为B和U。
6.3.6 导入结构参数
可以通过单击Structure parameters页面底部右边的Import…按钮,然后从文件选择对话框中,选择VESTA所支持的格式的文件,从存储结构数据的文件中导入结构数据。
选项Link指定以VESTA格式在文件*.vesta中输出当前结构数据的方式。结构数据通常记录在*.vesta中。另一方面,对于体数据,数据文件的相对路径(包括文件名)记录在*.vesta中,而不是在其中直接记录体数据。在勾选了Link选项时,也将使用数据文件的相对路径记录结构数据。然后,即使在保存*.vesta之后更改了结构数据文件,当重新打开*.vesta时,这些更改将反映在VESTA中。该选项在准备*.vesta文件时很有用,它以VESTA格式作为模板文件,用于设置对象和整体外观。此选项禁用Structure parameters选项卡中的文本框和列表框,从而禁止用户编辑结构数据,因为当启用此选项时,对结构数据的任何修改都不会保存到文件中。
6.3.7 移除冗余原子
当空间群变为对称性较高的空间群时,或者当单位晶胞变换为较小的单位晶胞时(见6.2.6),同一原子位置可能由两个或两个以上的位置生成。在这种情况下,可以通过单击Remove duplicate atoms按钮来删除冗余位置。然后,将打开如图所示的对话框,需输入将两个原子之间距离的阈值,将距离小于该值的两个原子视为同一个位点。
阈值以Å为单位输入。增加阈值也有一定意义,例如,可从计算模拟得到的大晶胞中提取出一个平均结构。
6.4 体数据
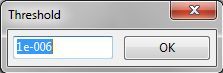
若要输入绘制有或无表面着色等值面的体数据,需单击Edit Data对话框中的Volumetric data选项卡(图6.13):
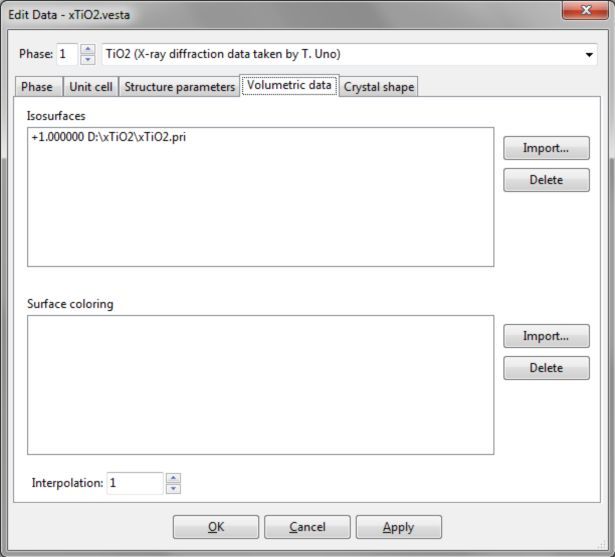
图6.13:Edit Data对话框中的Volumetric data选项卡
6.4.1 用于绘制等值面的体数据
可使用VESTA处理两个以上的体数据集。单击Import...在文件选择对话框中选择文件。在文件选择对话框中只有扩展名为所支持格式的文件是可见的。在选择体数据文件后,会出现Choose operations对话框(图6.14)。
图6.14:为体数据选择操作的对话框
操作
在Operation单选框中,除了数据单位转换外,还可以选择以下五个数据操作之一。
Add to current data与当前数据相加
Subtract from current data从当前数据中减去
Replace current data替换当前数据
Multiply to current data与当前数据相乘
Divide current data by new data用新数据除当前数据
利用Multiply to current data,可方便地利用波函数平方获得电子密度(电子的存在概率)。当前数据中不包含体数据时,不显示最后两个选项。在这种情况下,第一种和第三种选项的效果相同。
单位转换
在Convert the unit单选框中,数据的单位可由Å-3转换为bohr-3,反之亦然。选择Other factor选项,将数据乘以任意因子。然后,单击OK导入数据。重复上述步骤可以导入多个数据,如图6.15所示。
6.4.2 用于表面着色的体数据
用于表面着色的体数据可以与用于等值面绘制的数据以同样的方式导入。表面着色的一个典型应用是根据静电势对电子密度等值面进行着色。
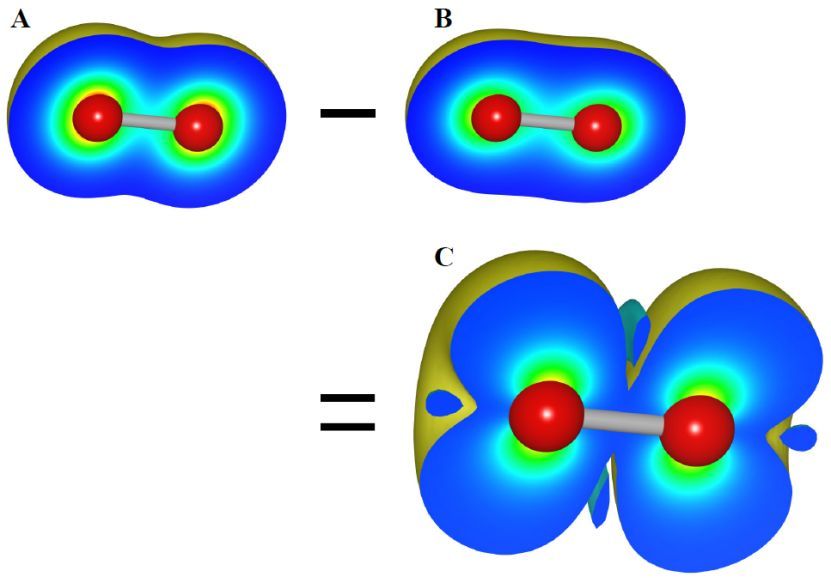
图6.15:O2分子中电子密度和有效自旋密度的分布。( A )自旋向上电子密度,ρ↑,( B )自旋向下电子密度,ρ↓和( C )用VESTA算得的有效自旋密度,Δρ =ρ↑-ρ↓。ρ↑和ρ↓都是用DVSCAT计算得到的。在( A )、( B )中,等值面设置为0.01a0-3,( C )中等值面设置为0.001a0-3,其中a0为玻尔半径
插值
利用三次样条插值算法可以提高体数据的空间分辨率。插值级别可在Interpolation文本框中设置。
6.5 晶体外形
可使用Edit Data对话框中的Crystal Shape选项卡(图6.16)输入和编辑晶体的外形。若要添加新的晶面,首先单击New按钮,输入Miller指数hkl和与原点(0, 0, 0)的距离。该距离可以以晶面间距d或Å为单位设置。晶面颜色和不透明度可设置为范围0 ~ 255的四个整数,或者使用颜色选择对话框和滑块设置,颜色选择对话框可通过单击文本框右侧的方形按钮打开。当勾选Apply symmetry operations选项时,将自动生成所有对称等价的晶面。
选择列表中的一项,单击Delete按钮,或按下键盘上的Delete键,可以删除一个晶面。单击Clear按钮,将删除列表中的所有晶面。
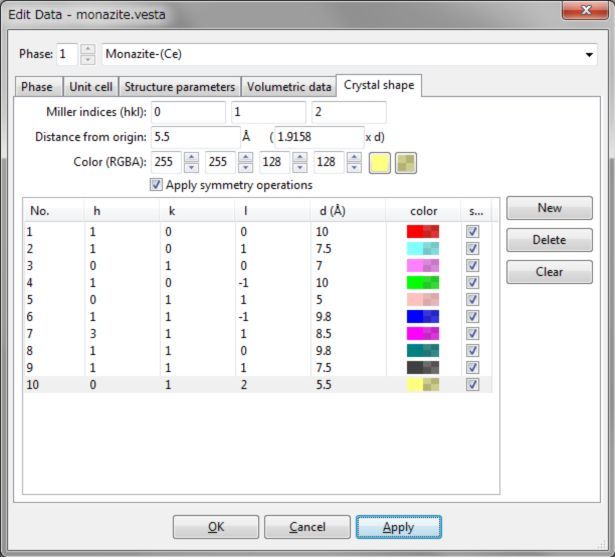
图6.16:Edit Data对话框中的Crystal Shape选项卡
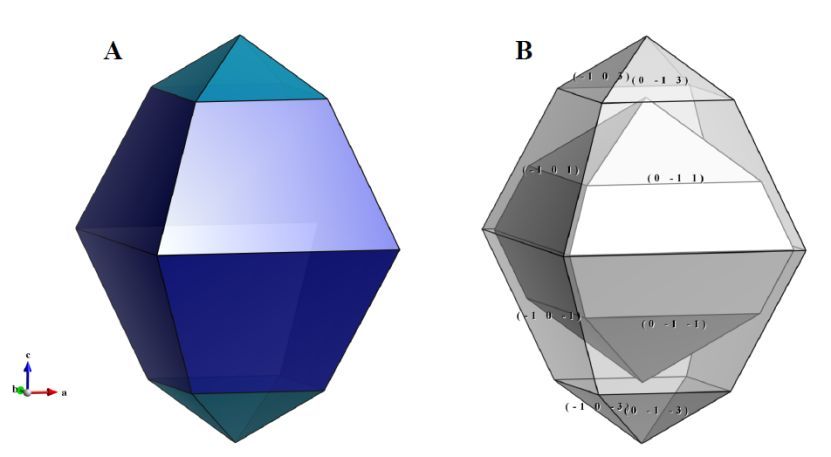
图6.17:锐钛矿相TiO2晶体的晶体形貌。在( B )中的内层晶体中,{101}和{103}晶面,离原点的距离均设为3.5 Å。在外层晶体中,{101}和{103}离原点的距离分别为3.8 Å和5 Å
6.5.1 示例
图6.17显示了由{101}面和{103}面组成的锐钛矿相TiO2晶体的形貌。为可视化晶体形貌,需确保指定的面组成一个封闭的多面体。例如,如果只为四方晶体指定{100},则只能形成沿c轴无限长的棱柱状形状。在这种情况下,图形区域中不可见任何对象。
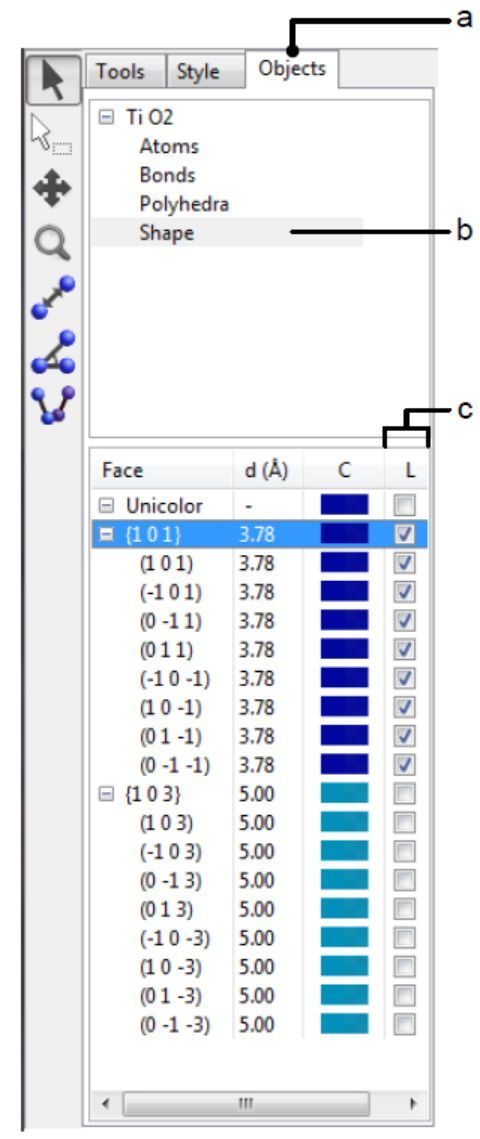
图6.18:侧边栏的Objects选项卡显示了晶面列表
要调整晶面的相对尺寸,可编辑从原点到晶面的距离。一般来说,晶面的相对面积随着距离的增加而减小,如图6.17B所示。当{103}面的距离设置为5 Å时,相对于{ 101 }面,这些面变得更小。晶体外形的原点与结构模型相同,即在晶体外形的中心放置一个分数坐标为(0, 0, 0)的位置。
当未勾选Apply symmetry operations选项时,可插入不遵循空间群对称性的晶面(图6.19A)。如果指定了相同米勒指数的多个晶面,与原点距离最小的晶面为可见(图6.19B、C)。这些特性使其可以表示真实晶体的晶习。
若要显示如图6.17B所示的晶面指数,需选择侧边栏中的Objects选项卡(图 6.18 , a),在页面上半部分的相列表中选择Shape(图 6.18 , b),然后单击标记为“L”(图 6.18 , c)的复选框。也可参见第12.2节中Objects选项卡的功能。
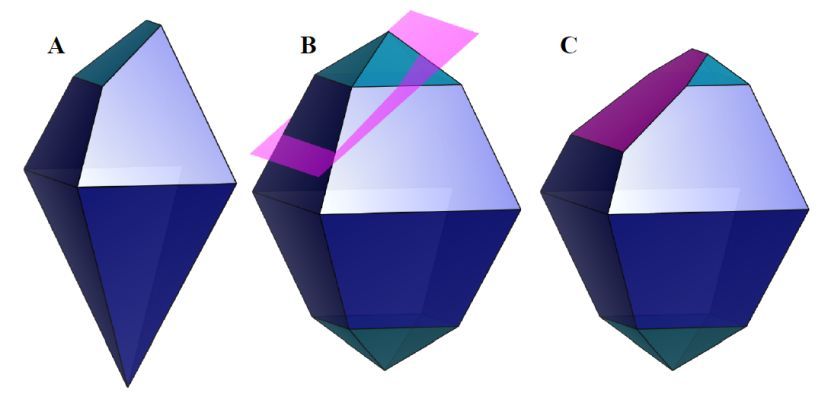
图6.19:锐钛矿相的晶体形貌,包含不遵循对称操作的晶面。( A )对于(-103)晶面不勾选Apply symmetry operations。( B )对于{101}和{103},与原点的距离分别设置为3.8 Å和5 Å,通过设置与原点的距离为3.8 Å插入(-103)晶面。( C )进一步添加了与( B )中的(-103)晶面重叠的 (-103)面

