
- 1linux性能工具--ftrace基础篇_linux trace函数
- 22.1 在Windows系统下载、安装、配置搭建Python开发环境——《跟老吕学Python》_redhat linux下python3.12.4开发环境配置
- 3Tomcat安全加固
- 4谷歌翻译国内使用_谷歌翻译镜像
- 5HTTP 请求中的 Content-Type 类型详解_content-type json
- 6飞凌嵌入式 gstreamer播放rtsp_嵌入式板卡gstreamer 可以播放网络上的视频吗
- 7LabView连接Access_labview连接access数据库
- 8[论文笔记] pai-megatron-patch Qwen2-72B/7B/1.5B 长文本探路
- 9Go语言:UUID 的生成与解析_uuid.newv4()
- 10开源项目介绍 |TKEStack-开源容器服务平台
生信入门(三)——使用limma、Glimma和edgeR,RNA-seq数据分析_limma makecontrasts
赞
踩
使用limma、Glimma和edgeR,RNA-seq数据分析
本篇继续上一篇数据预处理之后做差异表达分析。
六、差异表达分析
1、创建设计矩阵和对比
在此研究中,我们想知道哪些基因在我们研究的三组细胞之间以不同水平表达。我们的分析中所用到的线性模型假设数据是正态分布的。首先,我们要创建一个包含细胞类型以及测序泳道(批次)信息的设计矩阵。
#创建一个包含细胞类型和测序泳道信息的设计矩阵
design<-model.matrix(~0+group+lane)
colnames(design)<-gsub("group","",colnames(design))
- 1
- 2
- 3
design:

对于一个给定的实验,通常有多种等价的方法都能用来创建合适的设计矩阵。 比如说,~0+group+lane去除了第一个因子group的截距,但第二个因子lane的截距被保留。 此外也可以使用~group+lane,来自group和lane的截距均被保留。 理解如何解释模型中估计的系数是创建合适的设计矩阵的关键。 我们在此分析中选取第一种模型,因为在没有group的截距的情况下能更直截了当地设定模型中的对比。用于细胞群之间成对比较的对比可以在limma中用makeContrasts函数设定。
contr.matrix<-makeContrasts(
BasalvsLP=Basal-LP,
BasalvcML=Basal-ML,
LPvsML=LP-ML,
levels = colnames(design))
- 1
- 2
- 3
- 4
- 5
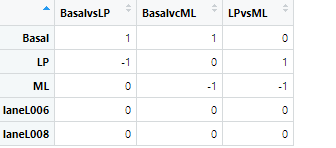
limma线性模型方法的核心优势之一便是其适应任意实验复杂程度的能力。简单的实验设计,比如此流程中关于细胞类型和批次的实验设计,直到更复杂的析因设计和含有交互作用项的模型,都能够被较简单地处理。当实验或技术效应可被随机效应模型(random effect model)模拟时,可以使用limma中的duplicateCorrelation函数来估计交互作用,这需要在此函数以及lmFit的线性建模步骤均指定一个block参数。
2、从表达计数数据中删除异方差
对于RNA-seq计数数据而言,原始计数或log-CPM值的方差并不独立于均值(Law et al. 2014)。有些差异表达分析方法使用负二项分布模型,假设均值与方差间具有二次的关系。而在limma中,假设log-CPM值符合正态分布,因此我们在对RNA-seq的log-CPM值进行线性建模时,需要使用voom函数计算每个基因的权重从而调整均值与方差的关系,否则分析得到的结果可能是错误的。
当操作DGEList对象时,voom从x中自动提取文库大小和归一化因子,以此将原始计数转换为log-CPM值。在voom中,对于log-CPM值额外的归一化可以通过设定normalize.method参数来进行。
下图左侧展示了这个数据集log-CPM值的均值-方差关系。通常而言,方差是测序实验操作中的技术差异和来自不同细胞类群的重复样本之间的生物学差异的结合,而voom图会显示出一个在均值与方差之间递减的趋势。 生物学差异高的实验通常会有更平坦的趋势,其方差值在高表达处稳定。生物学差异低的实验更倾向于急剧下降的趋势。
不仅如此,voom图也提供了对于上游所进行的过滤水平的可视化检测。如果对于低表达基因的过滤不够充分,在图上表达低的一端,受到非常低的表达计数的影响,会出现方差的下降。如果观察到了这种情况,应当回到最初的过滤步骤并提高用于该数据集的表达阈值。
当先前绘制的MDS图中发现组内重复样本的聚集与分离程度出现明显的差异时,可以用voomWithQualityWeights函数(Liu et al. 2015)来代替voom,在计算基因权重值以外还能计算每个样本的权重值。
par(mfrow=c(1,2))
v<-voom(x,design,plot=TRUE)
vfit<-lmFit(v,design)
vfit<-contrasts.fit(vfit,contrasts = contr.matrix)
efit<-eBayes(vfit)
plotSA(efit,main="Final model:Mean-variance trend")
- 1
- 2
- 3
- 4
- 5
- 6

- 图中绘制了每个基因的均值(x轴)和方差(y轴),显示了在该数据上使用voom前它们之间的相关性(左),以及当运用voom的权重后这种趋势是如何消除的(右)。左侧的图是使用voom函数绘制的,它为log-CPM转换后的数据拟合线性模型并提取残差方差。然后,对方差取四次方根(或对标准差取平方根),并相对每个基因的平均表达作图。均值通过平均计数加上2再进行log2转换计算得到。右侧的图使用plotSA绘制了log2残差标准差与log-CPM均值的关系。在这两幅图中,每个黑点表示一个基因。左侧图中,红色曲线展示了用于计算voom权重的估算所得的均值-方差趋势。右侧图中,由经验贝叶斯算法得到的平均log2残差标准差由水平蓝线标出。
3、拟合线性模型以进行比较
limma的线性建模使用lmFit和contrasts.fit函数进行,它们原先是为微阵列而设计的。这些函数不仅可以用于微阵列数据,也可以用于使用voom计算了基因权重后的RNA-seq数据。每个基因的表达值都会单独拟合一个模型。然后通过借用全体基因的信息来进行经验贝叶斯调整(empirical Bayes moderation),这样可以更精确地估算各基因的差异性(Smyth 2004)。图4为此模型的残差关于平均表达值的图。从图中可以看出,方差不再与表达水平均值相关。
4、检查DE基因数量
为快速查看表达差异水平,显著上调或下调的基因可以汇总到一个表格中。显著性的判断使用默认的校正p值阈值,即5%。在basal与LP的表达水平之间的比较中,发现了4648个在basal中相较于LP下调的基因,和4863个在basal中相较于LP上调的基因,即共9511个差异表达基因。在basal和ML之间发现了一共9598个差异表达基因(4927个下调基因和4671个上调基因),而在LP和ML中发现了一共5652个差异表达基因(3135个下调基因和2517个上调基因)。在basal类群所参与的比较中皆找到了大量的差异表达基因,这与我们在MDS图中观察到的结果相吻合。
summary(decideTests(efit))
- 1

在某些时候,不仅仅需要用到校正p值阈值,还需要差异倍数的对数(log-FCs)也高于某个最小值来更为严格地定义显著性。treat方法(McCarthy and Smyth 2009)可以按照对最小log-FC值的要求,使用经过经验贝叶斯调整的t统计值计算p值。当我们要求差异表达基因的log-FC显著大于1(等同于不同细胞类群之间表达量差两倍)并对此进行检验时,差异表达基因的数量得到了下降,basal与LP相比只有3648个差异表达基因,basal与ML相比只有3834个差异表达基因,LP与ML相比只有414个差异表达基因。
tfit<-treat(vfit,lfc = 1)
dt<-decideTests(tfit)
summary(dt)
- 1
- 2
- 3
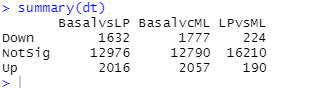
在多个对比中皆差异表达的基因可以从decideTests的结果中提取,其中的0代表不差异表达的基因,1代表上调的基因,-1代表下调的基因。共有2784个基因在basal和LP以及basal和ML的比较中都差异表达,其中的20个于下方列出。write.fit函数可用于将三个比较的结果一起提取并写入一个输出文件。
de.common<-which(dt[,1]!=0 & dt[,2]!=0)
length(de.common)
head(tfit$genes$SYMBOL[de.common],n=20)
vennDiagram(dt[,1:2],circle.col =c("turquoise","salmon"))
- 1
- 2
- 3
- 4
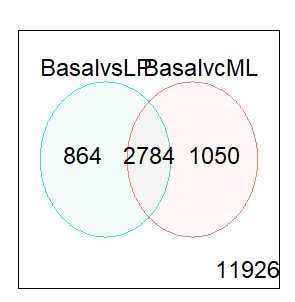
- 韦恩图展示了仅basal和LP(左)、仅basal和ML(右)的对比的DE基因数量,还有两种对比中共同的DE基因数量(中)。在任何对比中均不差异表达的基因数量标于右下。
write.fit(tfit,dt,file = "result.txt")#保存处理结果
- 1
5、检查单个DE基因
使用topTreat函数可以列举出使用treat得到的结果中靠前的DE基因(对于eBayes的结果则应使用topTable函数)。默认情况下,topTreat将基因按照经过多重假设检验校正的p值从小到大排列,并为每个基因给出相关的基因信息、log-FC、平均log-CPM、校正t值、原始及校正p值。列出前多少个基因的数量可由用户设定,如果设为n=Inf则会包括所有的基因。基因Cldn7和Rasef在basal与LP和basal于ML的比较中都位于DE基因的前几名。
basal.vs.lp<-topTreat(tfit,coef = 1,n=Inf)
basal.vs.ml<-topTreat(tfit,coef = 2,n=Inf)
head(basal.vs.lp)
head(basal.vs.ml)
- 1
- 2
- 3
- 4

6、差异表达结果可视化
为可视化地总结所有基因的结果,可使用plotMD函数绘制均值-差异关系(MD)图,其中展示了线性模型拟合所得到的每个基因log-FC与log-CPM平均值间的关系,而差异表达的基因会被重点标出。
limma:
#差异表达结果可视化
plotMD(tfit,column = 1,status = dt[,1],main=colnames(tfit)[1],xlim=c(-8,13))
- 1
- 2

Glimma的glMDPlot函数提供了交互式的均值-差异图,拓展了这种图表的功能。此函数的输出为一个html页面,左侧面板为结果的总结性图表(与plotMD的输出类似),右侧面板展示了各个样本的log-CPM值,而下方面板为结果的汇总表。这使得用户可以使用提供的注释中的信息(比如基因符号)搜索特定基因,而这在R统计图中是做不到的。
Glimma:
glMDPlot(tfit,coef = 1,status = dt,main = colnames(tfit)[1],side.main = "ENTREZID",counts = lcpm,groups = group,launch = TRUE)
- 1

使用Glimma生成的均值-差异图。左侧面板显示了总结性数据(log-FC与log-CPM值的关系),其中选中的基因在每个样本中的数值显示于右侧面板。下方为结果的表格,其搜索框使用户得以使用可行的注释信息查找某个特定基因,如基因符号Clu。
- Glimma提供的交互性使得单个图形窗口内能够呈现出额外的信息。 Glimma是以R和Javascript实现的,使用R代码生成数据,并在之后使用Javascript库D3(https://d3js.org)转换为图形,使用Bootstrap库处理界面并生成互动性可搜索的表格的数据表。这使得图表可以在任意当代浏览器中直接查看而无需后段服务器时刻运行,对于将其作为关联文件附加在Rmarkdown分析报告中而言非常方便。
前文所展示的图表中,一些仅展示了在任意一个条件下表达的所有基因而缺少单个基因的具体信息(比如不同对比中共同差异表达基因的韦恩图或均值-差异图),而另一些仅展示单个基因(交互式均值-差异图右边面板中所展示的log-CPM值)。而热图使用户得以查看某一小组基因的表达情况,既便于查看单个组或样本的表达,又不至于在关注于单个基因时失去对于整体的注意,也不会造成由于对上千个基因取平均值而导致的分辨率丢失。
使用gplots包的heatmap.2函数,我们为basal与LP的对照中前100个差异表达基因(按校正p值排序)绘制了一幅热图。此热图中正确地将样本按照细胞类型聚类,并重新排序了基因,表达相似的基因形成了块状。从热图中,我们发现basal与LP之间的前100个差异表达基因在ML和LP样本中的表达非常相似。
#gplots绘制热图
library(gplots)
basal.vs.lp.topgenes<-basal.vs.lp$ENTREZID[1:100]#取100个即可,没有对某一特征进行排序
i<-which(v$genes$ENTREZID %in% basal.vs.lp.topgenes)
mycol<-colorpanel(1000,"blue","white","red")
heatmap.2(lcpm[i,],scale = "row",labRow = v$genes$SYMBOL[i],labCol = group,
col = mycol,trace = "none",density.info = "none",margins = c(8,6),
lhei = c(2,10),dendrogram = "column")
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8

- 在basal和LP的对比中前100个DE基因log-CPM值的热图。经过缩放调整后,每个基因(每行)的表达均值为0,并且标准差为1。给定基因相对高表达的样本被标记为红色,相对低表达的样本被标记为蓝色。浅色和白色代表中等表达水平的基因。样本和基因已通过分层聚类的方法重新排序。图中显示有样本聚类的树状图。
7、使用camera的基因检验
在此次分析的最后,我们要进行一些基因集检验。为此,我们将camera方法(Wu and Smyth 2012)应用于Broad Institute的MSigDB c2中的(Subramanian et al. 2005)中适应小鼠的c2基因表达特征,这可从http://bioinf.wehi.edu.au/software/MSigDB/以RData对象格式获取。 此外,对于人类和小鼠,来自MSigDB的其他有用的基因集也可从此网站获取,比如标志(hallmark)基因集。C2基因集的内容收集自在线数据库、出版物以及该领域专家,而标志基因集的内容来自MSigDB,从而获得具有明确定义的生物状态或过程。
load(system.file("extdata", "mouse_c2_v5p1.rda", package = "RNAseq123"))
idx <- ids2indices(Mm.c2,id=rownames(v))
#Basal 与LP做对比
cam.BasalvsLP <- camera(v,idx,design,contrast=contr.matrix[,1])
head(cam.BasalvsLP,5)
#Basal 与ML做对比
cam.BasalvsML <- camera(v,idx,design,contrast=contr.matrix[,2])
head(cam.BasalvsML,5)
#LP与ML做对比
cam.LPvsML <- camera(v,idx,design,contrast=contr.matrix[,3])
head(cam.LPvsML,5)
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11

camera函数通过比较假设检验来评估一个给定基因集中的基因是否相对于不在集内的基因而言在差异表达基因的排序中更靠前。 它使用limma的线性模型框架,并同时采用设计矩阵和对比矩阵(如果有的话),且在检验的过程中会运用到来自voom的权重值。 在通过基因间相关性(默认设定为0.01,但也可通过数据估计)和基因集的规模得到方差膨胀因子(variance inflation factor),并使用它调整基因集检验统计值的方差后,将会返回根据多重假设检验进行了校正的p值。
绘制LP与ML的条码图
#绘制条码图
barcodeplot(efit$t[,3], index=idx$LIM_MAMMARY_LUMINAL_MATURE_UP,
index2=idx$LIM_MAMMARY_LUMINAL_MATURE_DN, main="LPvsML")
- 1
- 2
- 3

今日告一段落啦,明天见,加油呀~

