
- 13D-DIC数字图像相关法测量流程介绍-数字图像采集_视觉软件中触发周期是什么意思
- 2【windows】亲测-win11系统跳过联网和微软账户登录,实现本地账户登录_win11跳过联网激活
- 3【数据结构和算法初阶(C语言)】时间复杂度(衡量算法快慢的高端玩家,搭配例题详细剖析)_衡量一个算法好坏一般以最坏的时间复杂度为标准
- 4如何在群晖NAS搭建bitwarden密码管理软件并实现无公网IP远程访问_群晖怎么安装bitwarden
- 5基于Hadoop的区块链海量数据存储的设计与实现_区块链 hdfs
- 6iOS(一):Swift纯代码模式iOS开发入门教程_swift 开发ios入门教程
- 7学懂C语言系列(三):C语言基本语法
- 8kafka架构深入
- 9Langchain-chatchat: Langchain核心组件及应用_langchain chatchat
- 10【爬虫】1.4 POST 方法向网站发送数据_网页爬虫 post数据
基于人工智能(AI)的蛋白结构预测工具合集
赞
踩
王建民
蛋白质折叠涉及重新排列空间中的线性氨基酸序列,使其处于低能状态。仅基于氨基酸序列预测正确的三维结构非常困难,其原因在于肽键的自由组合会产生大量的可能性:即使对于一个小的蛋白质进行结构域顺序抽样,耗费的时间都将超过宇宙的年龄,因此计算预测是为了规避顺序抽样问题。在过去的40年里,计算预测一直在稳步改进,通过X 射线晶体学、核磁共振波谱和低温电子显微镜(cryo-EM)等实验确定蛋白质结构序列的同源性。自1994年起,每两年通过将计算预测方法应用于最新解析的蛋白质结构评估其性能。三年前,AlphaFold的首次实施已经是将人工智能应用于解析蛋白质结构的革命性进步。2020年,重新设计的AlphaFold近乎完美地精确预测出了蛋白质三维结构,其预测水准与实验室水平相差无几,一举破解了困扰学界长达五十年之久的“蛋白质折叠”难题。DeepMind团队提出了一种计算方法名为AlphaFold 2,即使在不知道相似结构的情况下,也可以以原子精度定期预测蛋白质结构。2021年7月15日,DeepMind团队在Nature杂志上发表了文章"Highly accurate protein structure prediction with AlphaFold",描述了AlphaFold2是一个基于神经网络的全新设计的AlphaFold版本,其预测的蛋白质结构能达到原子水平的准确度。2021年7月15日,华盛顿大学蛋白设计研究所David Baker教授课题组及其他合作机构在Science上发表论文"Accurate prediction of protein structures and interactions using a three-track neural network",公布了其开源蛋白质预测工具RoseTTAFold的研究结果。
AlphaFold2 和 RoseTTAFold 等数据和人工智能驱动的蛋白质折叠预测工具为大分子结构预测和设计提供了强大的驱动力。本文旨在总结相关工具。
https://github.com/sacdallago/folding_tools
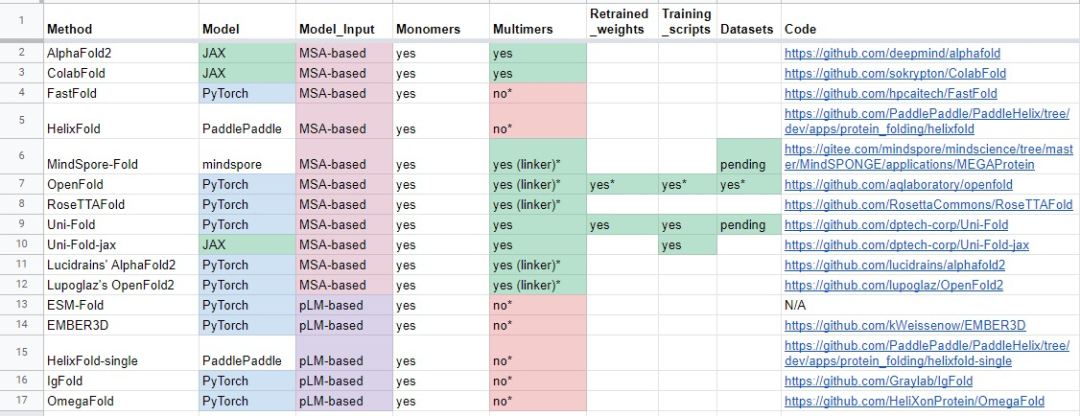
声明:本文非商业目的,图片主要来源于twitter,google和微信群聊,部分文字摘自工具简介。
基于 MSA
使用多序列比对 (MSA) 作为输入
AlphaFold2
2021年7月15日,DeepMind团队在Nature杂志上发表了文章"Highly accurate protein structure prediction with AlphaFold",描述了AlphaFold2是一个基于神经网络的全新设计的AlphaFold版本,其预测的蛋白质结构能达到原子水平的准确度。目前,DeepMind 在 GitHub 上公开了 AlphaFold2 的源代码,还免费开放了 AlphaFold 数据集。
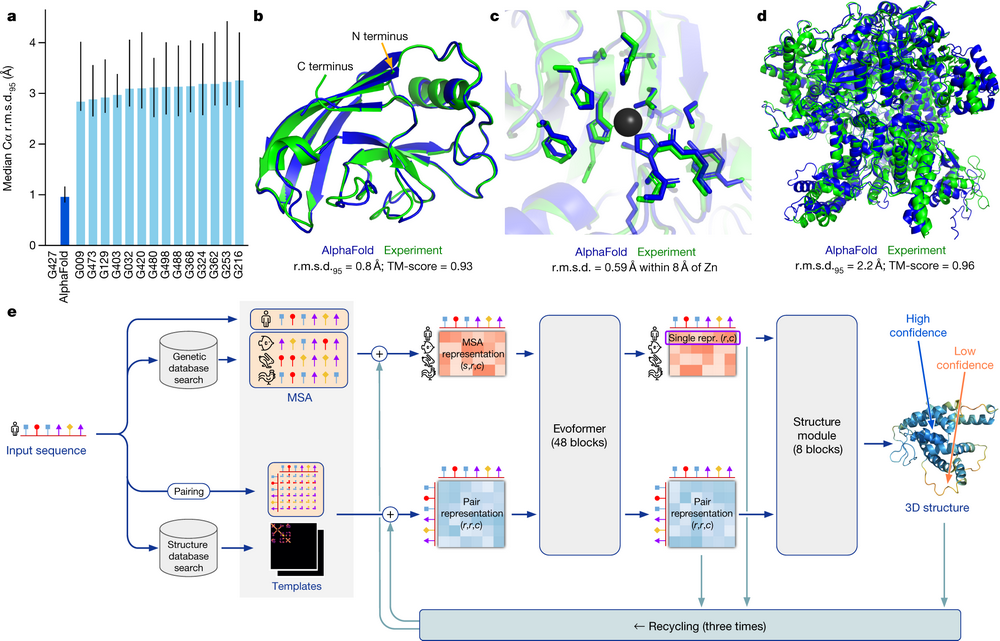
https://github.com/deepmind/alphafold
https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebooks/AlphaFold.ipynb
https://alphafold.ebi.ac.uk/
Jumper, J., Evans, R., Pritzel, A. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
https://doi.org/10.1038/s41586-021-03819-2
RoseTTAFold
2021年7月15日,华盛顿大学蛋白设计研究所David Baker教授课题组及其他合作机构在Science上发表论文"Accurate prediction of protein structures and interactions using a three-track neural network",公布了其开源蛋白质预测工具RoseTTAFold的研究结果。
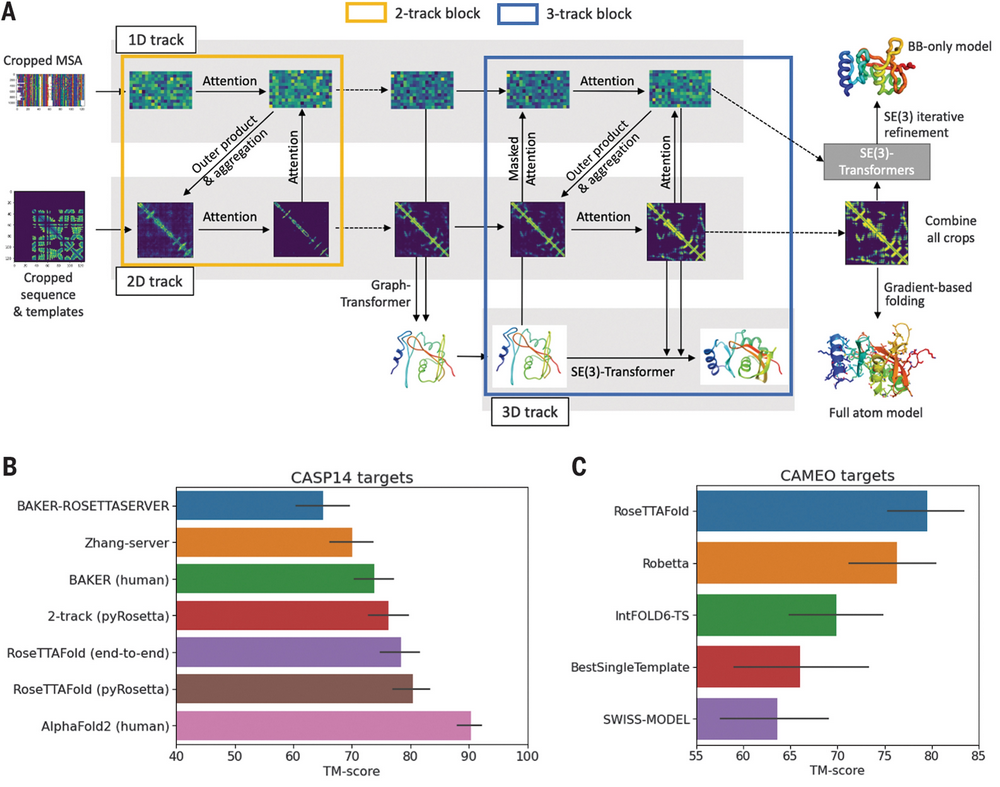
https://github.com/RosettaCommons/RoseTTAFold
https://www.ipd.uw.edu/2021/07/rosettafold-accurate-protein-structure-prediction-accessible-to-all/
https://robetta.bakerlab.org/
https://www.rosettacommons.org/docs/latest/application_documentation/structure_prediction/fold-and-dock
Accurate prediction of protein structures and interactions using a three-track neural network. Science 15 Jul 2021:eabj8754
DOI: 10.1126/science.abj8754
ColabFold
ColabFold 通过将 MMseqs2 的快速同源搜索与 AlphaFold2 或 RoseTTAFold 相结合,加速预测蛋白质结构和复合物。ColabFold 的搜索速度提高了 40-60 倍,并优化了模型利用率,每天可以在具有一个图形处理单元的服务器上预测近 1,000 个结构。与 Google Colaboratory 相结合,ColabFold 成为一个免费且可访问的蛋白质折叠平台。
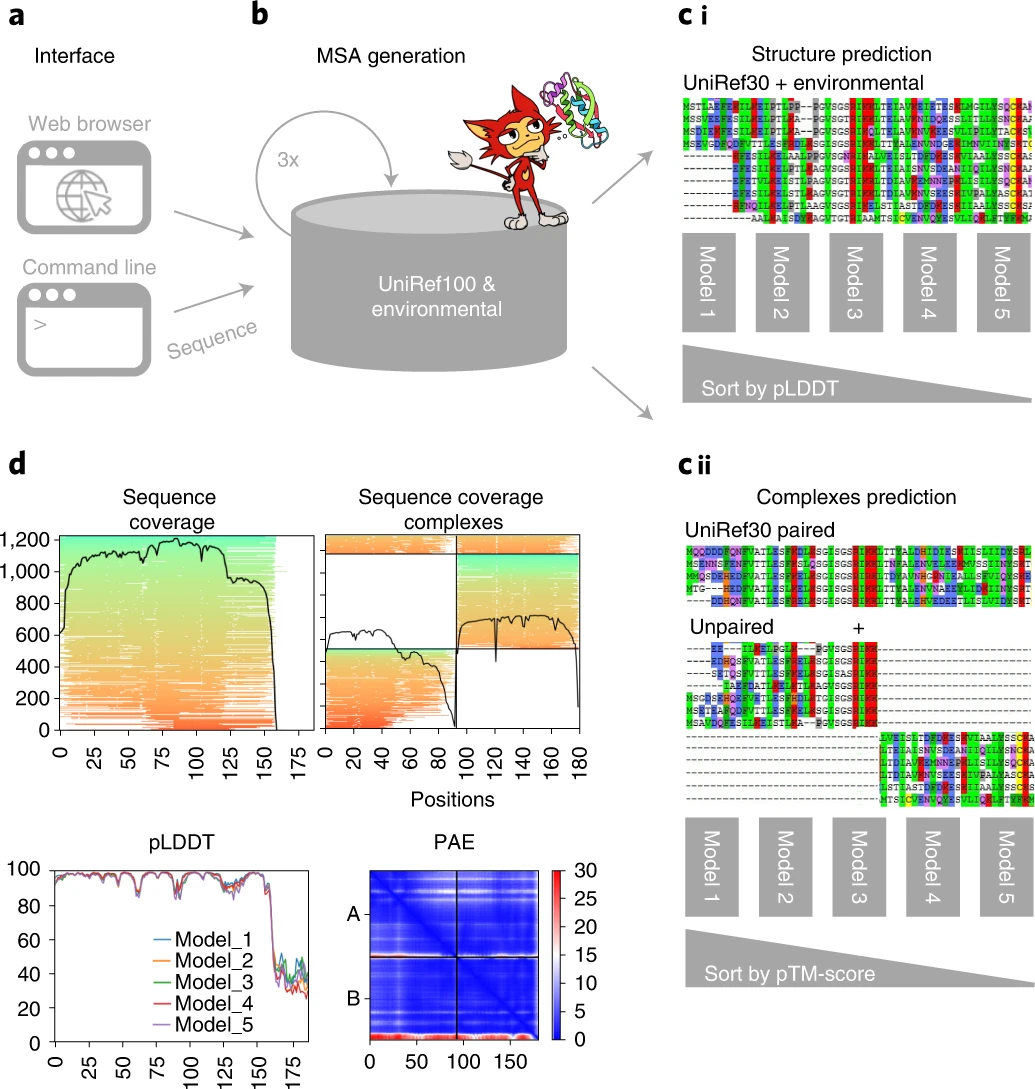
https://github.com/sokrypton/ColabFold
https://colabfold.mmseqs.com/
Mirdita, M., Schütze, K., Moriwaki, Y. et al. ColabFold: making protein folding accessible to all. Nat Methods 19, 679–682 (2022).
https://doi.org/10.1038/s41592-022-01488-1
OpenFold
OpenFold 是 DeepMind 的 AlphaFold2 的 PyTorch 复现版本,用于自动处理蛋白质折叠实验。OpenFold 并不是同类中的第一个,但它是迄今为止最完整的,并且拥有与 AlphaFold 相等或更高的能力。与 AlphaFold 一样,以自己的名义,OpenFold 是完全开源的,并且在非常宽松的许可下提供。两者的参数都可以轻松下载并在 CC BY 4.0 下获得许可,而通过 GitHub 提供的代码在 Apache 2.0 下获得许可。这意味着任何有兴趣的人都可以将 OpenFold 用于几乎任何目的。
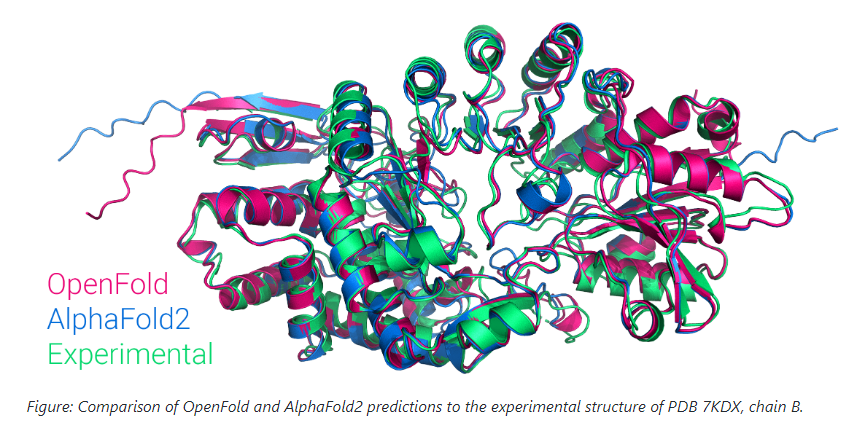
OpenFold 和 AlphaFold 之间最明显的区别在于,虽然 AlphaFold 是为 JAX 工作流开发的,但 OpenFold 的所有代码都基于 PyTorch 环境。OpenFold 也是可训练的,这意味着可以为专业研究创建变体,这与 AlphaFold 不同。
https://openfold.io/
https://github.com/aqlaboratory/openfold
https://colab.research.google.com/github/aqlaboratory/openfold/blob/main/notebooks/OpenFold.ipynb
https://huggingface.co/nz/OpenFold
Uni-Fold
深势科技重磅推出蛋白质结构预测工具 Uni-Fold,成功复现曾引起生物学界轰动的 AlphaFold2 的全规模训练,并开源训练代码与推理代码。
Uni-Fold 克服了 AlphaFold2 未开源训练代码、硬件支持单一、模型不可商用等局限性,在训练与推理环节进行了 NVIDIA GPU上的适配、性能优化及功能完善等工作,为更多人参与推动领域进一步发展提供了基础。
https://github.com/dptech-corp/Uni-Fold
https://colab.research.google.com/github/dptech-corp/Uni-Fold/blob/main/notebooks/unifold.ipynb
Li, Z., Liu, X., Chen, W., Shen, F., Bi, H., Ke, G. and Zhang, L., 2022. Uni-Fold: An Open-Source Platform for Developing Protein Folding Models beyond AlphaFold. bioRxiv.
FastFold
蛋白质结构预测是结构生物学领域理解基因翻译和蛋白质功能的重要方法。AlphaFold 将 Transformer 模型引入了具有原子精度的蛋白质结构预测领域。然而,AlphaFold 模型的训练和推理由于其特殊的性能特点和巨大的内存消耗,既耗时又昂贵。在本文中,作者提出了 FastFold,这是一种用于训练和推理的蛋白质结构预测模型的高效实现。FastFold 包括一系列基于对 AlphaFold 性能的全面分析的 GPU 优化。同时,通过 Dynamic Axial Parallelism 和 Duality Async Operation,FastFold 实现了高模型并行缩放效率,超越现有流行的模型并行技术。实验结果表明,FastFold 将整体训练时间从 11 天减少到 67 小时,并实现了 7.5-9.5 倍的长序列推理加速。此外,我们将 FastFold 扩展到 512 个 GPU,并以 90.1% 的并行效率实现了总计 6.02 PetaFLOPs。

https://github.com/hpcaitech/FastFold
Cheng, S., Wu, R., Yu, Z., Li, B., Zhang, X., Peng, J. and You, Y., 2022. FastFold: Reducing AlphaFold Training Time from 11 Days to 67 Hours. arXiv preprint arXiv:2203.00854.
HelixFold
百度飞桨强大的高性能并行计算能力支持下,飞桨螺旋桨 PaddleHelix 生物计算团队发布了蛋白结构预测模型 HelixFold,围绕着显存峰值、训练速度、分布式策略进行了全面性能优化。通过与原版 AlphaFold2 模型和哥伦比亚大学 Mohammed AlQuraishi 教授团队基于 PyTorch 复现的 OpenFold 模型的性能对比测试显示,HelixFold 模型的训练性能相比 AlphaFold2 提升106.97%,相比 OpenFold 提升104.86%。
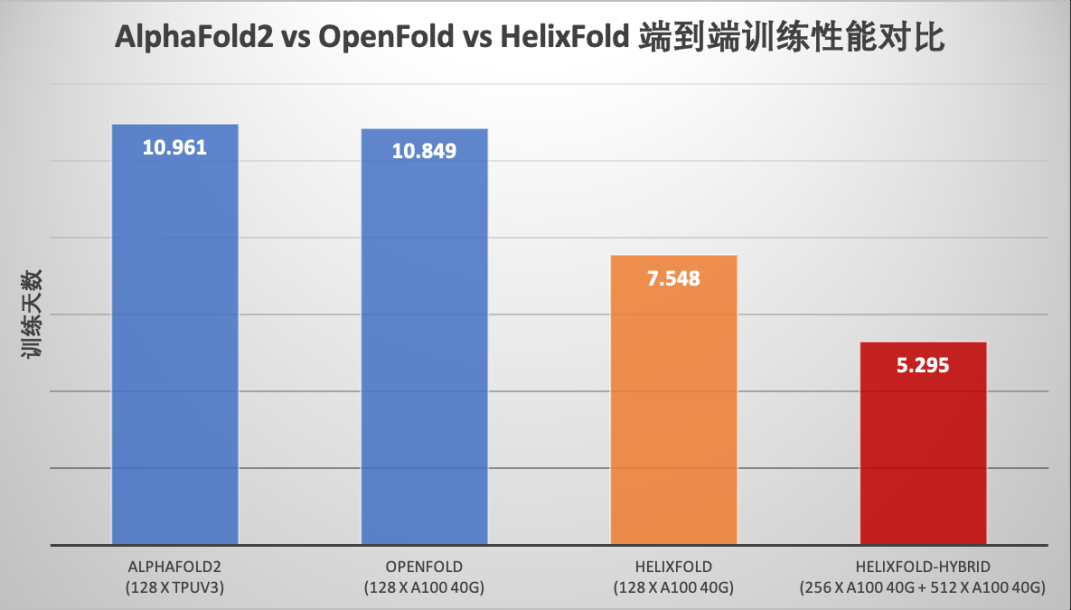
https://github.com/PaddlePaddle/PaddleHelix/tree/dev/apps/protein_folding/helixfold
Wang, G., Fang, X., Wu, Z., Liu, Y., Xue, Y., Xiang, Y., Yu, D., Wang, F. and Ma, Y., 2022. HelixFold: An Efficient Implementation of AlphaFold2 using PaddlePaddle. arXiv preprint arXiv:2207.05477.
MEGA-Fold
使用计算机高效计算获取蛋白质空间结构的过程被称为蛋白质结构预测,传统的结构预测工具一直存在精度不足的问题,直至2020年谷歌DeepMind团队提出AlphaFold2,该模型相较于传统工具预测精度大幅提升,所得结构与真实结构误差接近实验方法,但是仍存在数据前处理耗时过长、缺少MSA时预测精度不准、缺乏通用评估结构质量工具的问题。针对这些问题,高毅勤老师团队与MindSpore科学计算团队合作进行了一系列创新研究,开发出更准确和更高效的蛋白质结构预测工具MEGA-Protein。
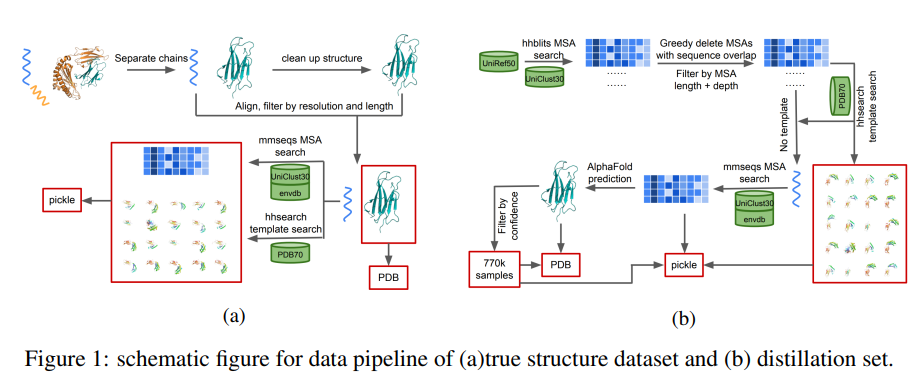
MEGA-Protein主要由三部分组成:蛋白质结构预测工具MEGA-Fold,MSA生成工具MEGA-EvoGen和蛋白质结构评分工具MEGA-Assessement。
https://gitee.com/mindspore/mindscience/tree/master/MindSPONGE/applications/MEGAProtein
Liu, S., Zhang, J., Chu, H., Wang, M., Xue, B., Ni, N., Yu, J., Xie, Y., Chen, Z., Chen, M. and Liu, Y., 2022. PSP: Million-level Protein Sequence Dataset for Protein Structure Prediction. arXiv preprint arXiv:2206.12240.
基于 pLM
使用来自蛋白质语言模型 (pLM) 的嵌入作为输入
ESM-Fold
大型语言模型超越简单的模式匹配来执行更高级别的推理并生成逼真的图像和文本。虽然在较小规模上研究了针对蛋白质序列训练的语言模型,但随着规模的扩大,人们对它们对生物学的了解知之甚少。在这项工作中,研究人员训练了多达 150 亿个参数的模型,这是迄今为止要评估的最大的蛋白质语言模型。研究人员发现,随着模型的缩放,它们学习的信息能够以单个原子的分辨率预测蛋白质的三维结构。研究人员提出了 ESMFold,用于直接从蛋白质的单个序列进行高精度的端到端原子级结构预测。ESMFold 与 AlphaFold2 和 RoseTTAFold 对于语言模型可以很好理解的低困惑度序列具有相似的准确性。ESMFold 推理比 AlphaFold2 快一个数量级,从而能够在实际时间尺度上探索宏基因组蛋白的结构空间。
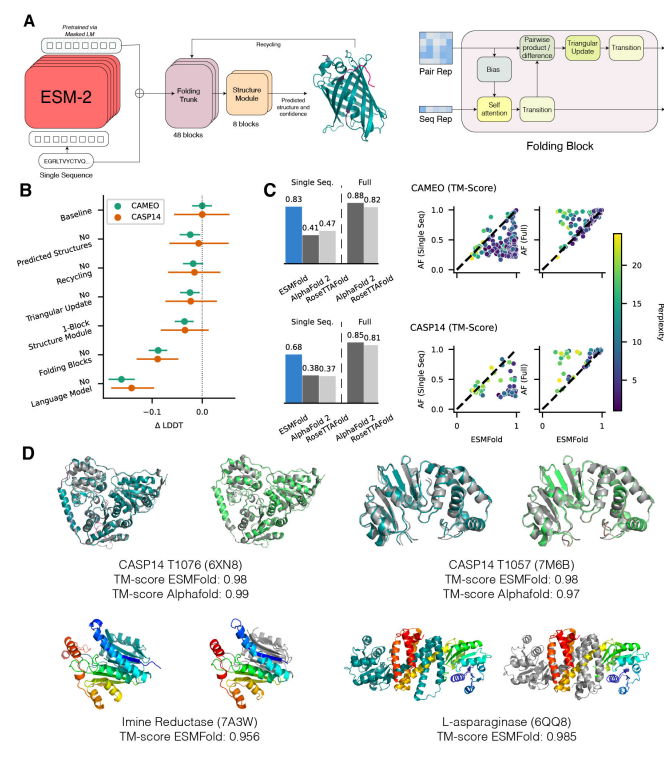
Lin, Z., Akin, H., Rao, R., Hie, B., Zhu, Z., Lu, W., dos Santos Costa, A., Fazel-Zarandi, M., Sercu, T., Candido, S. and Rives, A., 2022. Language models of protein sequences at the scale of evolution enable accurate structure prediction. bioRxiv.
HelixFold-single
通过人工智能的方法,从蛋白质的一级序列精准预测其三维结构,已被AlphaFold2证实可达到实验精度。但AlphaFold2的模型主要依赖多序列比对(MSA)和模版(Template)信息,而从蛋白质数据库中搜索MSA和模版又是一件非常耗时的工作,成为模型向产业界大规模推广的一个瓶颈。HelixFold-Single将自然语言处理领域的语言模型,应用在蛋白质上,构建了预训练语言模型,从3亿的无标注蛋白质数据中提取信息,建模蛋白质之间的关系,从而将MSA同源信息隐式的学习在语言模型中,进而有效地替代MSA信息检索模块,极大地提升了结构预测的速度。在精度不输AlphaFold2的基础上,效率最多提升近千倍 。
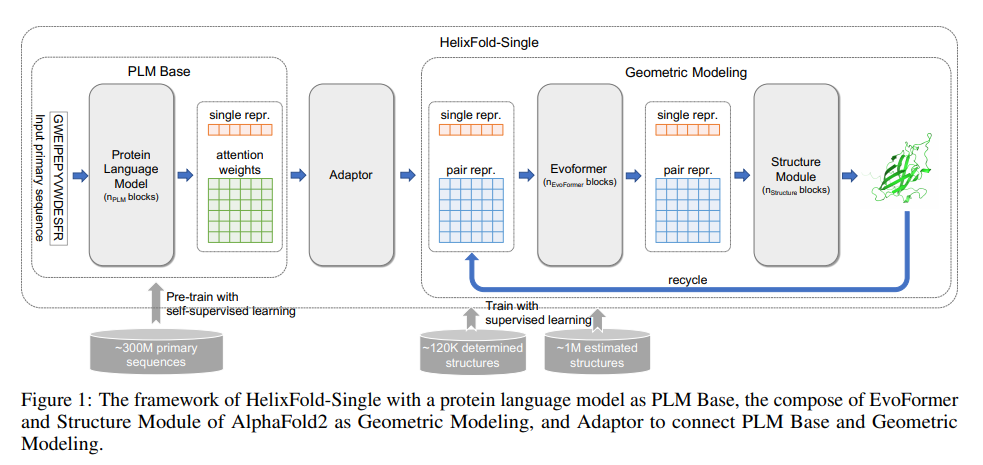
https://github.com/PaddlePaddle/PaddleHelix/tree/dev/apps/protein_folding/helixfold-single
Fang, X., Wang, F., Liu, L., He, J., Lin, D., Xiang, Y., Zhang, X., Wu, H., Li, H. and Song, L., 2022. HelixFold-Single: MSA-free Protein Structure Prediction by Using Protein Language Model as an Alternative. arXiv preprint arXiv:2207.13921.
OmegaFold
AI预测蛋白质3D结构,仅通过单条蛋白序列就能搞定。也就是说,AI预测蛋白质结构,可以不需要蛋白质进化过程中的同源信息。一些人工设计的蛋白质药物和工业合成用酶,也可以通过AI预测3D结构,确定其对人体的功能。
达成这一成就的AI算法名为OmegaFold,在最近的全球持续蛋白质预测竞赛中,整体预测能力已经与DeepMind此前开发的AlphaFold2,和华盛顿大学开发的RoseTTAFold不相上下,甚至有些指标优于后两者。
研究成果来自国内AI创新药物公司华深智药,一家成立不到一年的中国初创企业,孵化自张亚勤旗下的清华大学智能产业研究院。
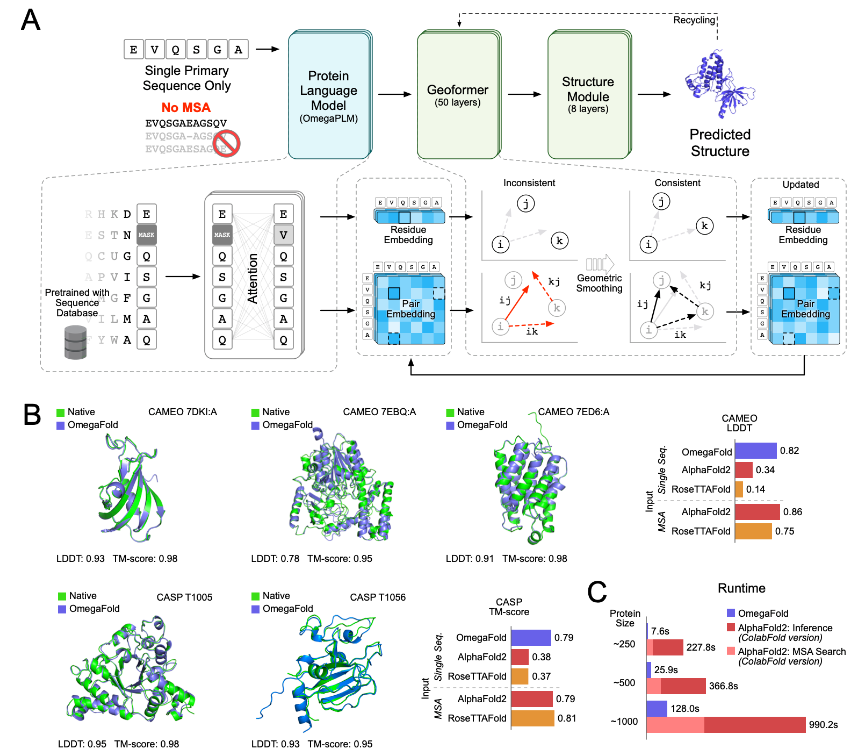
https://github.com/HeliXonProtein/OmegaFold
https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/beta/omegafold.ipynb
Wu, R., Ding, F., Wang, R., Shen, R., Zhang, X., Luo, S., Su, C., Wu, Z., Xie, Q., Berger, B. and Ma, J., 2022. High-resolution de novo structure prediction from primary sequence. bioRxiv.
IgFold
抗体的结合是由一组6个loops的结构决定,这个6个loops构成互补决定族(CDR)。这些CDR loops的精准建模可以深入了解这些结合机制和提高使特定抗体的合理设计成为可能。
5个CDR loops倾向于采用通过序列相似性可以有效地进行预测,然而,重链的第3个CDR loop(CDR H3),由于其在序列和长度上的增加的多样性,不能通过序列相似性有效的预测,已被证明是对模型是很大的挑战。重链和轻链之间界面上的H3 loops的位置,使其构象依赖于链间方向(inter-chain orientation),鉴于H3 loop在结合中的核心作用,预测H3 loops结构的效果对于了解抗体-抗原相互作用,实现抗体的合理设计非常重要。
JOHNS HOPKINS大学发表了可以快速预测抗体结构的IgFold深度学习方法,其准确率可以与AlphaFold2媲美。IgFold先从一个预训练的语言模型提取预序列表征(该模型在558M自然抗体序列上进行的训练),然后通过图网络直接预测backbone atoms坐标。其预测结构质量与其他工具类似或更好(包括AlphaFold),且预测所需时间不到1分钟。在这个时间尺度上进行准确的结构预测,使得以前不可行的调研与验证方法成为可能。
同时为了证明IgFold的能力,作者预测了105K配对的8个抗体序列的结构,将观察到的抗体结构空间扩大了40倍以上。
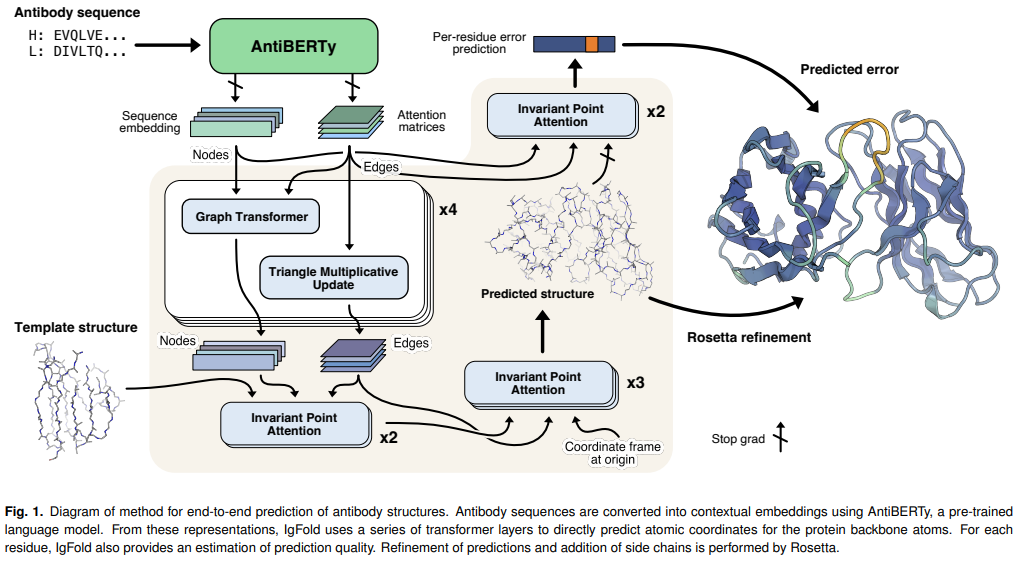
https://github.com/Graylab/IgFold
https://colab.research.google.com/github/Graylab/IgFold/blob/main/IgFold.ipynb
Ruffolo, J.A. and Gray, J.J., 2022. Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies. Biophysical Journal, 121(3), pp.155a-156a.
参考资料
Jumper, J., Evans, R., Pritzel, A. et al. Highly accurate protein structure prediction with AlphaFold. Nature (2021).
https://doi.org/10.1038/s41586-021-03819-2
Accurate prediction of protein structures and interactions using a three-track neural network. Science 15 Jul 2021:eabj8754
DOI: 10.1126/science.abj8754
Edich, M., Briggs, D.C., Gao, Y., Kippes, O. and Thorn, A., 2022. The impact of AlphaFold on experimental structure solution. Faraday Discussions.
https://github.com/sacdallago/folding_tools
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集





