
热门标签
热门文章
- 1计算机网络中23是什么端口号,什么是网络端口,端口有哪些分类-电脑自学网
- 2【话题】程序员应该有什么职业素养
- 3java的JVM学习总结第一章——JVM与java的体系结构
- 4【文献阅读】受山体阴影影响的冰湖制图方法研究(Li JunLi等人,2018.09,IJRS)_阴影损失文献
- 5【数据结构】线性表之《带头双向循环链表》超详细实现
- 6LLM 大模型学习必知必会系列(十):基于AgentFabric实现交互式智能体应用,Agent实战_dashscope中的llm的api
- 7ue4 UMG添加到场景_unreal umg放到世界空间
- 8译文:dBA和dBC的不同_dba和dbc的区别
- 9网站高性能架构设计——web前端与池化_前端架构设计
- 10spring整合openAI大模型之Spring AI_spring ai maven
当前位置: article > 正文
GEO数据库学习二(提取表达矩阵 ID转换准备)_gpl6244
作者:人工智能uu | 2024-07-07 00:19:55
赞
踩
gpl6244
上一节记录了如何下载数据,这一节学习id转换,主要是探针对应到symbol,其中表达矩阵的行是探针id,列是样品id,ids的列是探针id和symbol,我们的目的是利用探针id,将二者对应起来。
1.如何从对象提取表达矩阵
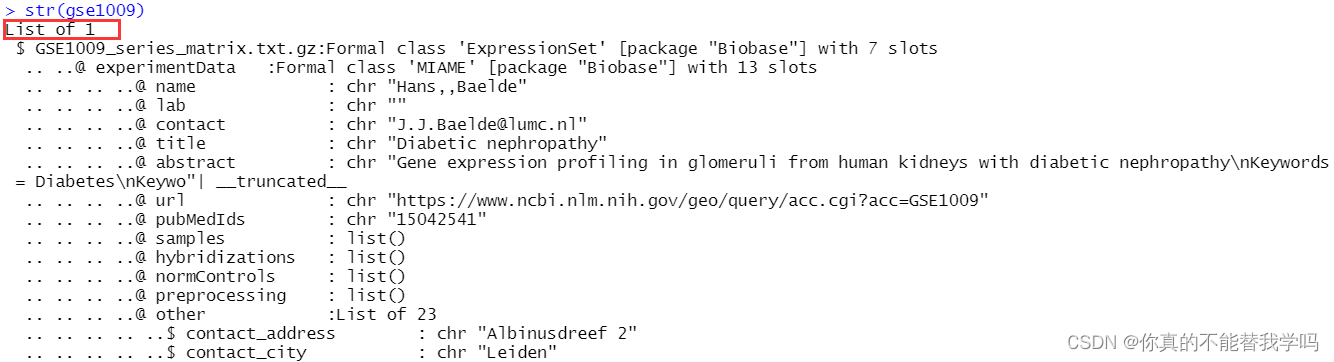
按照上一篇文章getGEO方式下载下来的GSE是一个对象,数据类型是list,里面存放着很多信息。
利用str(gse1009) 可以发现这是一个列表,则可以用gse1009[[1]] 查看第一列。
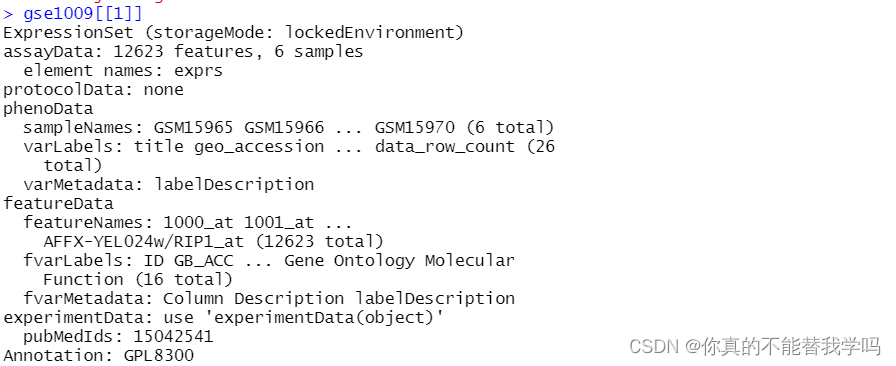
本篇id转换利用的是GSE42872,采用上一节getGSE的方法下载数据。
gse42872 <- getGEO('GSE42872', destdir=".",GSEMatrix=T,AnnotGPL=F)
#从gse42872对象中提取表达矩阵
a=gse42872[[1]]
View(a)
b=exprs(a)
View(b)
- 1
- 2
- 3
- 4
- 5
- 6
- 7
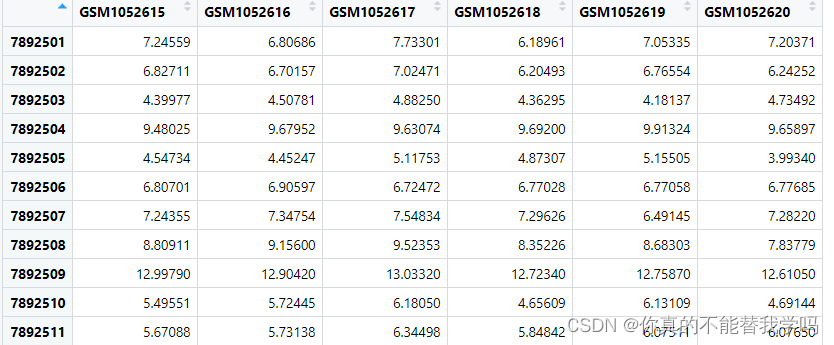
2.ID转换
得到的b的行名就是探针id,需要转换成基因id,需要注意的是每一种芯片平台都有不同的包来转换,利用gse42872[[1]] 查看后的最后一行:Annotation: GPL6244就是这个数据的芯片平台,再到官网或者用R获取芯片探针与基因的对应关系三部曲-bioconductor里可以查看对应的包,这里的GPL6244对应的是:hugene10sttranscriptcluster。
提取表达矩阵:
BiocManager::install("hugene10sttranscriptcluster.db") library("hugene10sttranscriptcluster.db") ids=toTable(hugene10sttranscriptclusterSYMBOL) View(ids) #unique()去除重复 查看这一列不重复的项 也就是查看有多少基因 length(unique(ids$symbol)) #查看最后几个基因涉及到的探针数目 #table(ids$symbol)是将基因名那一列转为表格统计数目 #(sort(table(ids$symbol))是排序 这样就能知道一个基因对应几个探针 tail(sort(table(ids$symbol))) #查看所有基因涉及到的探针数目 可以知道大多数基因都对应一个探针 table(sort(table(ids$symbol))) #画图查看 plot(table((sort(table(ids$symbol)))))

- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19

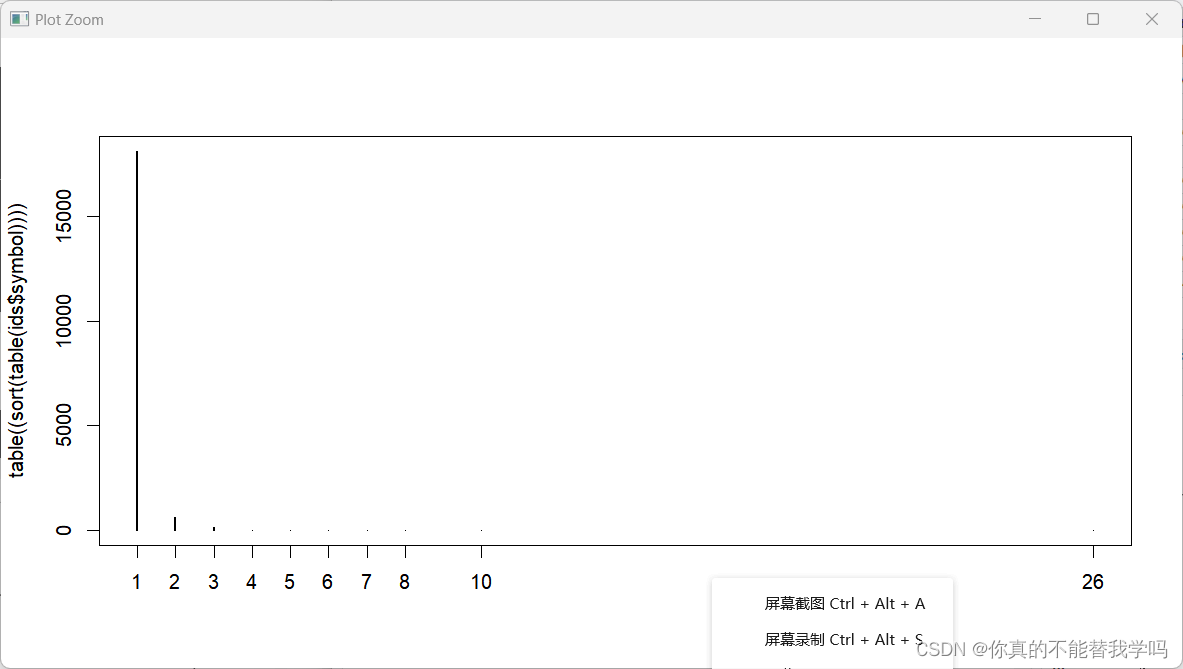
id转换
> exprSet=b > #检查b的探针有多少在ids表里 > #%in%是匹配的意思 判断左边的元素是否等于右边 > table(rownames(exprSet) %in% ids$probe_id) FALSE TRUE 13443 19854 > #只取存在于ids表里的基因所对应的探针行 > exprSet=exprSet[rownames(exprSet) %in% ids$probe_id,] > dim(exprSet) [1] 19854 6 > #ids表里面也只保留表达矩阵exprSet里有的 > ids=ids[ids$probe_id %in% rownames(exprSet)] Error in `[.data.frame`(ids, ids$probe_id %in% rownames(exprSet)) : 选择了未定义的列 > ids=ids[rownames(exprSet) %in% ids$probe_id] Error in `[.data.frame`(ids, rownames(exprSet) %in% ids$probe_id) : 选择了未定义的列 > #ids表里面也只保留表达矩阵exprSet里有的 > #因为表达矩阵已经经过筛选 则和ids里行数不一样 此时需要选择match函数来匹配 > ids=ids[match(rownames(exprSet),ids$probe_id),] > head(ids) probe_id symbol 1 7896746 MTND1P23 2 7896754 SEPTIN7P13 3 7896759 LINC01128 4 7896761 SAMD11 5 7896779 KLHL17 6 7896798 PLEKHN1 > exprSet[1:5,1:5] GSM1052615 GSM1052616 GSM1052617 GSM1052618 GSM1052619 7896746 10.16530 10.50290 10.13550 10.69020 10.34200 7896754 8.81485 8.99721 9.19503 9.49915 9.86029 7896759 8.75126 8.61650 8.81149 8.32067 8.41445 7896761 8.39069 8.52617 8.43338 9.17284 9.10216 7896779 8.20228 8.30886 8.18518 8.13322 8.06453
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
表达矩阵分类:
如果有一个基因名对应多个探针,就会得到一个新的探针列表:
#表达矩阵分类 #by()分类汇总 依次是data indices(因子或因子列表) 函数 #rowMeans(x) 行平均值 即某个探针对应的表达量 #which.max()输出最大值对应的序号 #rownames(x)输出最大值对应的序号对应的行名即探针 > tmp =by(exprSet, + ids$symbol, + function(x) rownames(x)[which.max(rowMeans(x))]) #probes新的探针列表 > probes=as.character(tmp) #表达矩阵过滤前的维度 > dim(exprSet) [1] 19854 6 > exprSet=exprSet[rownames(exprSet) %in% probes,] > dim(exprSet) [1] 18851 6
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
参考文章:GEO数据库视频学习笔记(ID转换)
声明:本文内容由网友自发贡献,不代表【wpsshop博客】立场,版权归原作者所有,本站不承担相应法律责任。如您发现有侵权的内容,请联系我们。转载请注明出处:https://www.wpsshop.cn/w/人工智能uu/article/detail/794371
推荐阅读
相关标签

