
- 1java获取ad域组信息_AD工作室精心研发漏洞安全扫描器
- 2Mamba2-minimal(mamba2最简实现)的使用与若干问题解决办法_mamba2 代码解析
- 3html5 xdwlnjs cn,最近需要调用一个网站的js,但是发现是加密的,有大佬来解密下吗?...
- 4【协议详解】PON/EPON/GPON/OAM/OMCI基本原理(2024最新/最全)_ploam协议分析功能
- 5基于MATLAB的说话人识别系统
- 6Linux系统配置核查-【等保测评】网络安全等级保护测评 S3A3 计算环境操作系统(Linux)_等保 s3a3_等保测评 liunx未提供设置敏感标记功能。
- 7springboot实现数据脱敏(重写序列化方法)_spring boot数据脱敏form表单没生效
- 8Git撤销对远程仓库的commit、push操作
- 9spark RPC详解_spark rpc很高
- 10无线网破解 跑字典 EWSA使用教程
GEO数据挖掘-PCA、差异分析_如何分析geo表达数据
赞
踩
From 生物技能树 GEO数据挖掘第二节
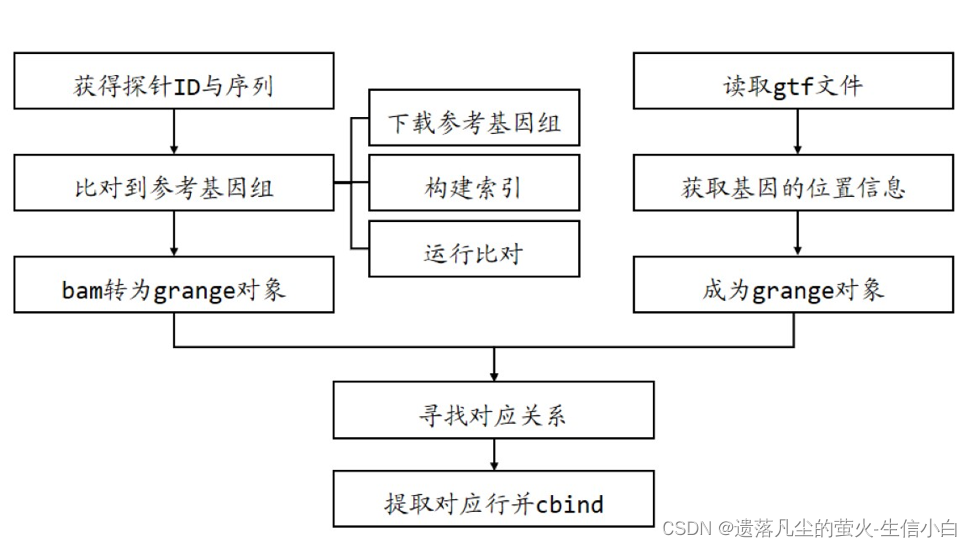
探针注释
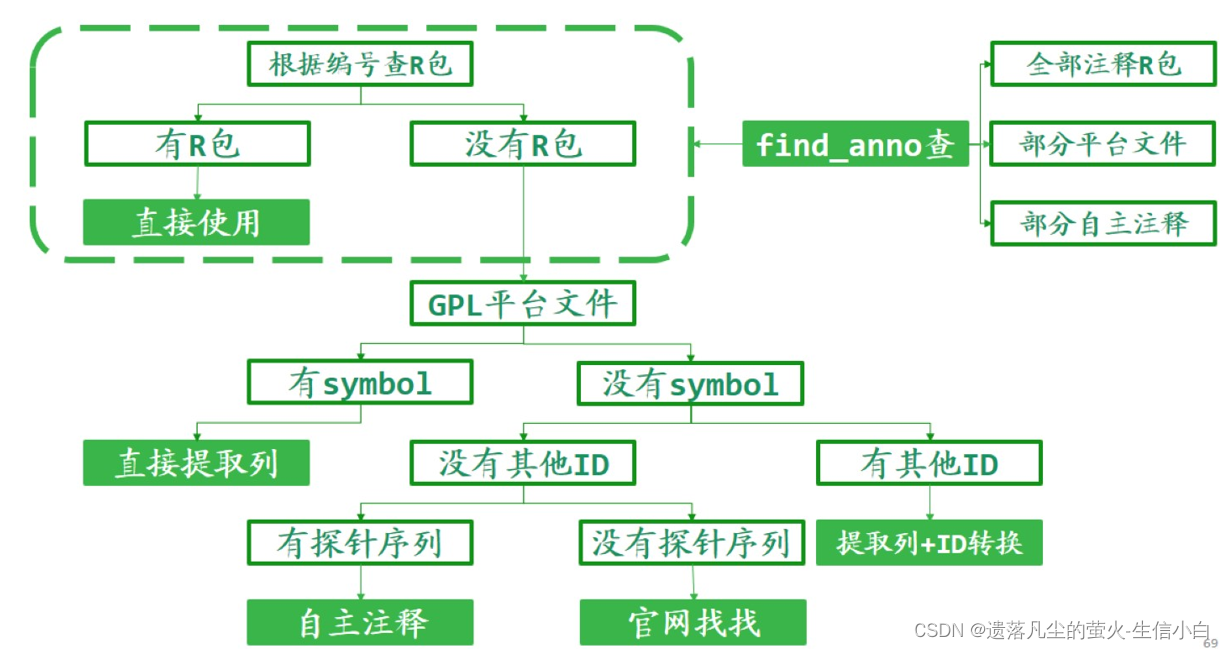
自主注释流程(了解)
rm(list = ls())
load(file = "step2output.Rdata")
#输入数据:exp和Group
- 1
- 2
- 3
PCA图、top1000基因热图
探针注释
查看示例代码
示例代码:
# The variable Species (index = 5) is removed
# before PCA analysis
iris.pca <- PCA(iris[,-5], graph = FALSE)
fviz_pca_ind(iris.pca,
geom.ind = "point", # show points only (nbut not "text")
col.ind = iris$Species, # color by groups
palette = c("#00AFBB", "#E7B800", "#FC4E07"),
addEllipses = TRUE, # Concentration ellipses
legend.title = "Groups"
)
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
dat=as.data.frame(t(exp))
#根据sthda上的示例数据,更改自己的数据,需要转置后转为dataframe
- 1
- 2
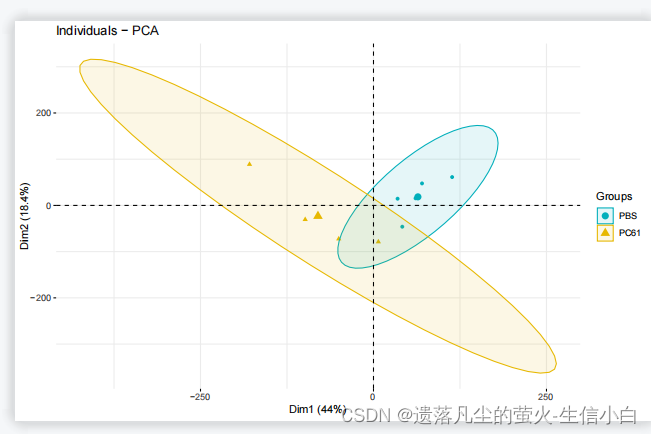
#画PCA图,使用pca分析后的数据dat.pca
如果每组样本在四个以下,是不会有圈的,圈是置信区间,在统计学里,小于4个样本没法计算置信区间
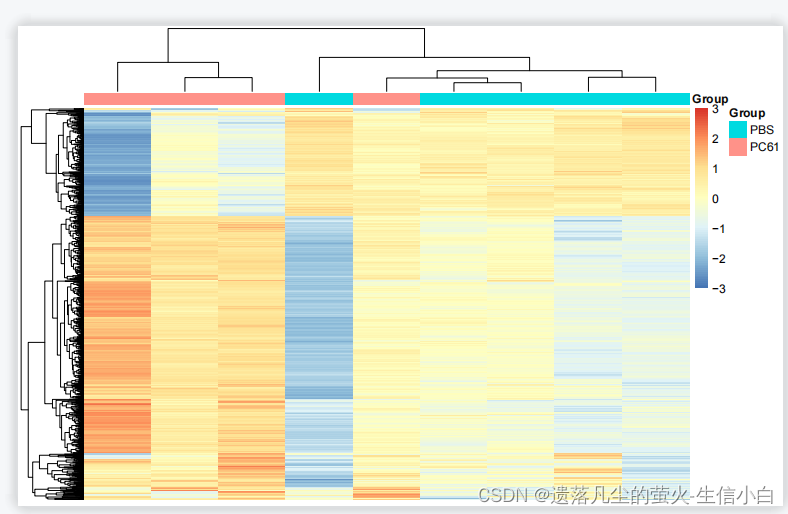
top 1000 sd 热图
离散基因热图,top1000表达基因,只是看一下,不用放文章里
g = names(tail(sort(apply(exp,1,sd)),1000)) #day7-apply的思考题,apply(exp,1,sd)对exp的每一行,就是一个基因,求sd,sort排序,小-到排序后,取后1000个,再提取基因名字(向量名字)
n = exp[g,]
library(pheatmap)
annotation_col = data.frame(row.names = colnames(n),
Group = Group)
pheatmap(n,
show_colnames =F,
show_rownames = F,
annotation_col=annotation_col,
scale = "row", #对数据进行转换,按行标准化,只保留行内差别,不保留行间差别,会把数据范围缩放到大概-5~5之间
breaks = seq(-3,3,length.out = 100) #设置色带分布范围为-3~3之间,超出此范围的数字显示极限颜色
)
# ?pheatmap,查看帮助文档
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
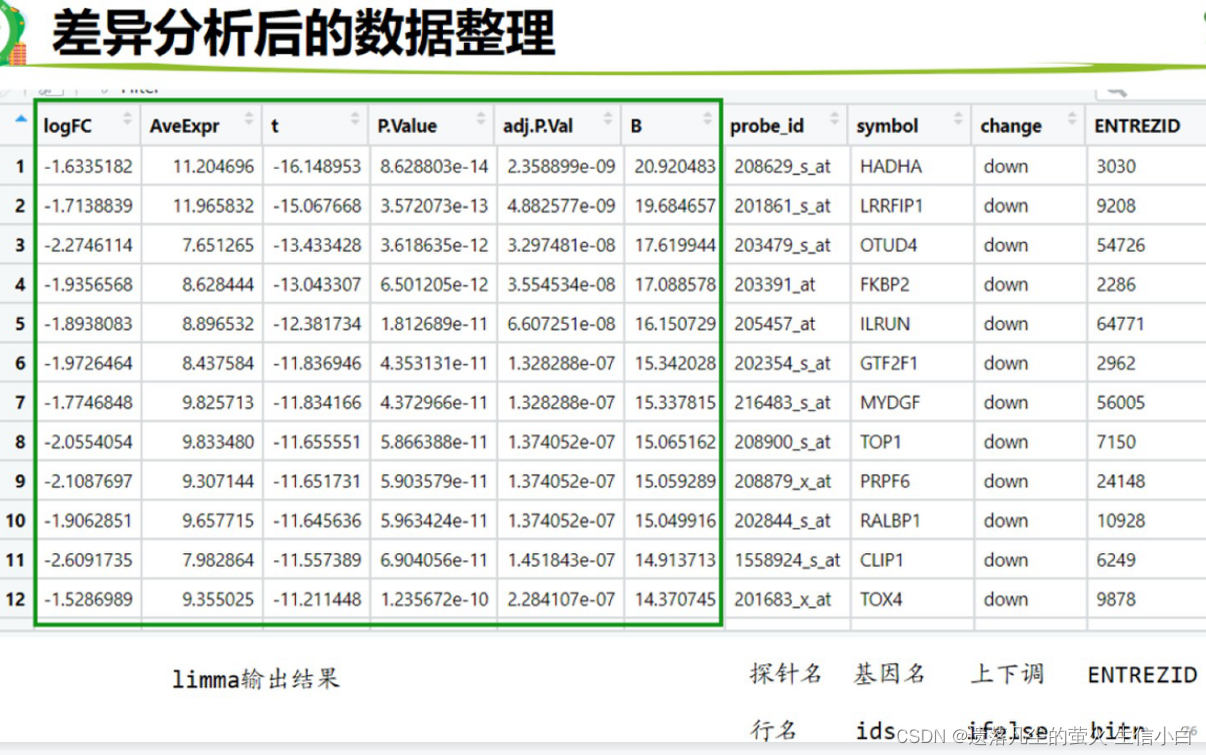
差异分析
1.得到差异基因结果表格,2.做了火山图,3.做热土图
rm(list = ls())
load(file = "step2output.Rdata")
#load上一步做完后得到用于差异分析的结果表格
#需要输入数据:exp、ids、group,使用limma包根据贝叶斯检验原理进行差异分析
library(limma)
design = model.matrix(~Group)
#model.matrix()根据分组信息生成一个模型矩阵,线性拟合函数需要用模型矩阵
fit = lmFit(exp,design)
#线性拟合函数,当你执行 fit = lmFit(exp, design) 这行代码时,你正在尝试拟合一个线性模型到表达数据 exp,其中:
exp:这是一个矩阵或数据框,包含了基因表达水平的测量值。每一行通常代表一个基因,每一列代表一个样本或实验条件。
design:这是一个设计矩阵,用于指定模型中每个样本的实验条件。它通常是一个因子向量或指示变量矩阵,用于定义模型中的各项,比如不同的处理组或时间点。
fit = eBayes(fit)
#贝叶斯检验
deg = topTable(fit,coef = 2,number = Inf)
#提取差异分析结果:coef = 2指design的第二列,number = infinity指提取全部的差异基因
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
得到一个probe_id和对应的logFC、pValue的表格,但还需要和symbol还有entrezid连接在一起
代码如下:
1.加probe_id列,把行名变成一列,防止行名丢失
library(dplyr)
deg = mutate(deg,probe_id = rownames(deg))
#mutate新增一列,probe_id = rownames(deg):这里创建了一个新的列 probe_id,其值是 deg 的行名。rownames() 函数用于获取数据框的行名,注意不能直接添加
- 1
- 2
- 3
2.加上探针注释
probe_id和基因symbol不是一对一的关系,是多对一或者一对多的关系,因此需要去重
- 一个探针对应多个基因:非特异性探针,在表格中可以看到,则需要直接去除
- 一个基因对应多个探针:相当于一个基因测了好多遍
处理方法:随机去重/保留行和或行平均值最大的探针/取多个探针的平均值
ids = distinct(ids,symbol,.keep_all = T)
#此处是随机去重的方法,其他去重方式在zz.去重方式.R
deg = inner_join(deg,ids,by="probe_id")
#用inner_join取交集并把差异分析的结果deg和之前的id—symbol表格连接在一起
nrow(deg)
## [1] 20824
#检查一下,如果行数为0就是你找的探针注释是错的。
- 1
- 2
- 3
- 4
- 5
- 6
- 7
#保留最大值
exp2 = exp[ids$probe_id,]
identical(ids$probe_id,rownames(exp2))
library(dplyr)
ids = ids %>%
mutate(exprowsum = rowSums(exp2)) %>%
arrange(desc(exprowsum)) %>%
select(-3) %>%
distinct(symbol,.keep_all = T)
nrow(ids)
# 拿这个ids去inner_join
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
#求平均值
rm(list = ls())
load("step2output.Rdata")
exp3 = exp[ids$probe_id,]
rownames(exp3) = ids$symbol
exp3[1:4,1:4]
exp4 = limma::avereps(exp3)
# 此时拿到的exp4已经是一个基因为行名的表达矩阵,直接差异分析,不再需要inner_join
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
3.加change列,标记上下调基因
logFC_t = 1 p_t = 0.05 #设置logFC和pValue的阈值 #使用ifelse两次或者casewhen判断down、up、stable并输出成新的一列change k1 = (deg$P.Value < p_t)&(deg$logFC < -logFC_t) k2 = (deg$P.Value < p_t)&(deg$logFC > logFC_t) deg = mutate(deg,change = ifelse(k1,"down",ifelse(k2,"up","stable"))) table(deg$change) ## ## down stable up ## 579 19621 624 #思考:如何使用padj而非p值 把代码里的所有的P.Value替换成adj.P.Val > colnames(deg) [1] "logFC" "AveExpr" "t" "P.Value" "adj.P.Val" "B" [7] "probe_id" "symbol"

- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
火山图
library(ggplot2)
ggplot(data = deg, aes(x = logFC, y = -log10(P.Value))) +
geom_point(alpha=0.4, size=3.5, aes(color=change)) +
scale_color_manual(values=c("blue", "grey","red"))+
geom_vline(xintercept=c(-logFC_t,logFC_t),lty=4,col="black",linewidth=0.8) +
geom_hline(yintercept = -log10(p_t),lty=4,col="black",linewidth=0.8) +
theme_bw()
- 1
- 2
- 3
- 4
- 5
- 6
- 7
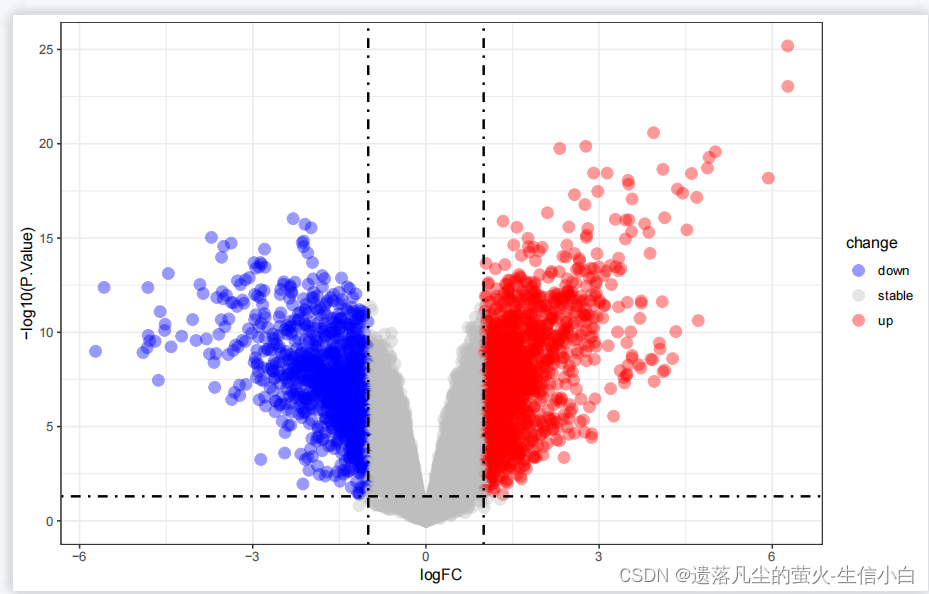
#使用ggplot的geom_point画火山图,vline和hline画阈值的线
差异基因热图
# 表达矩阵行名替换为基因名,分为两步:
exp = exp[deg$probe_id,]
#按deg中的symbol列的内容在exp中按行取子集,把最终使用的探针取出来
rownames(exp) = deg$symbol
#把exp中行名改为deg的symbol列,此时已经是一一对应的,上述俩代码只能运行一次,运行一次直接就把探针表达矩阵转换为基因表达矩阵了
diff_gene = deg$symbol[deg$change !="stable"]
#取出change列不是stable的基因symbol
n = exp[diff_gene,]
#按有差异的基因symbol在exp中按行取子集,即为有差异的基因的logFC和pValue,赋值到数据框n中,用于画差异基因热图
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
library(pheatmap)
annotation_col = data.frame(group = Group)
rownames(annotation_col) = colnames(n)
pheatmap(n,show_colnames =F,
show_rownames = F,
scale = "row",
#cluster_cols = F,
#即不按照列聚类,此时按照表达矩阵的顺序聚类
annotation_col=annotation_col,
breaks = seq(-3,3,length.out = 100)
)
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
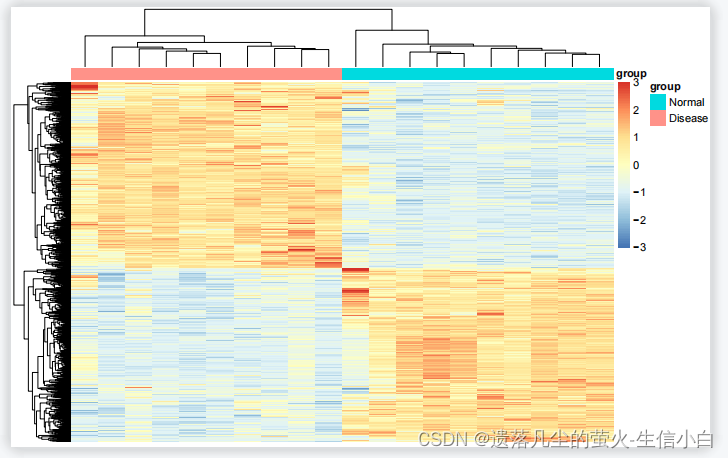
#如果差异基因的聚类分组还是错乱的,则加cluster_col = F
#如果加了还是错乱的,去看:小洁老师的语雀/分组聚类的热图
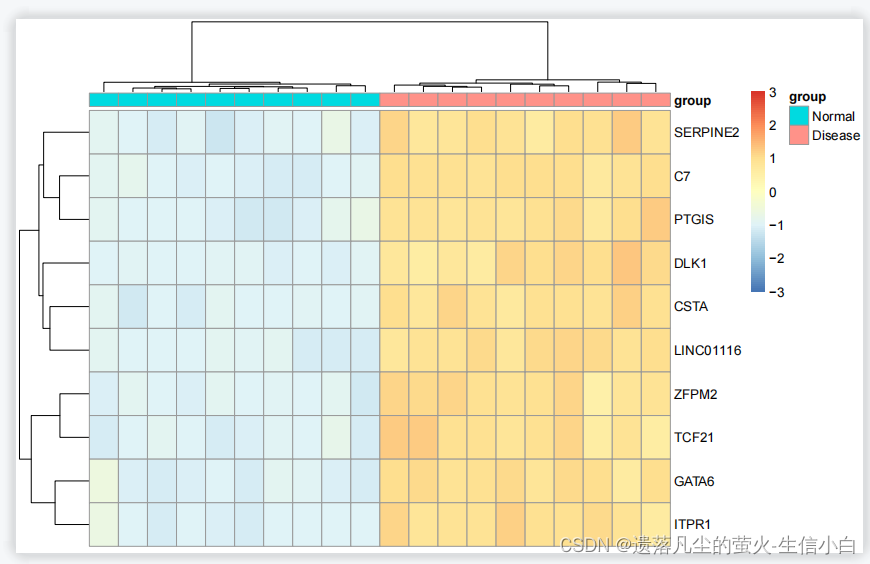
添加链接描述
#如果行名比较少,例如改成10行,就显示出基因
pheatmap(n[1:10,],show_colnames =F,
#show_rownames = F,
scale = "row",
#cluster_cols = F,
annotation_col=annotation_col,
breaks = seq(-3,3,length.out = 100)
)
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
转换行名的快捷函数:探针矩阵如何转换为基因矩阵
> library(tinyarray)
> exp[1:4,1:4]
GSM175766 GSM175767 GSM175768 GSM175769
1007_s_at 8.045017 8.314098 8.342717 8.261483
1053_at 6.444243 6.330321 6.168972 6.422393
117_at 6.158540 5.805438 5.565754 6.082891
121_at 7.737116 7.640965 7.835118 7.631916
> exp2 = trans_array(exp,ids)
20824 rownames transformed after duplicate rows removed
> exp2[1:4,1:4]
GSM175766 GSM175767 GSM175768 GSM175769
DDR1 8.045017 8.314098 8.342717 8.261483
RFC2 6.444243 6.330321 6.168972 6.422393
HSPA6 6.158540 5.805438 5.565754 6.082891
PAX8 7.737116 7.640965 7.835118 7.631916
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
转换id
symbol:常说的基因名
entrezid:富集分析指定用
两个并非一一对应,损失或增加部分基因属于正常,两者可以转换
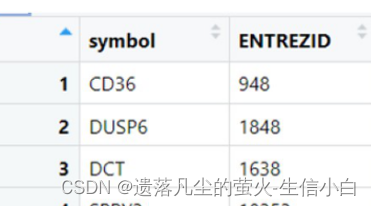
加ENTREZID列,用于富集分析(symbol转entrezid,然后inner_join)
entrezid是富集分析指定用的,需要symbol转entrezid,然后inner_join
使用clusterProfiler中的bitr函数实现,另外数据库根据物种不同
library(clusterProfiler)
library(org.Hs.eg.db)
s2e = bitr(deg$symbol,
fromType = "SYMBOL",
toType = "ENTREZID",
OrgDb = org.Hs.eg.db)#人类,注意物种,不同物种R包不同,如果物种写错,**也不会报错**,所以要检查代码错了没
- 1
- 2
- 3
- 4
- 5
- 6
一部分基因没匹配上是正常的。<30%的失败都没事。
其他物种http://bioconductor.org/packages/release/BiocViews.html#___OrgDb nrow(deg)
添加链接描述
看剩下数量,如果只有几十说明有问题
deg = inner_join(deg,s2e,by=c("symbol"="SYMBOL"))
#把差异基因的表和entrezid通过inner_join连接在一起,用于后面的富集分析
- 1
- 2
多了几行少了几行都正常,SYMBOL与ENTREZID不是一对一的
nrow(deg) #检查
## [1] 20827
#再看看还有几行,然后保存
save(exp,Group,deg,logFC_t,p_t,file = "step4output.Rdata")
- 1
- 2
- 3
- 4
富集分析-KEGG数据库
KEGG(Kyoto Encyclopedia of Genes and Genomes)是系统分析基因功能、基因组 信息数据库,它有助于研究者把基因及表达信息作为一个整体网络进行研究,以“理解生物系统的高级功能和实用程序资源库”著称。
补充
两个文件夹里分析出来的差异基因如何改交集
# 首先保存第一个project里的差异基因
exp = exp[deg$probe_id,]
rownames(exp) = deg$symbol
diff_gene = deg$symbol[deg$change !="stable"]
diff_gene2 = diff_gene
save(diff_gene2,file = "lianxi_diff_gene.Rdata")
#取交集
exp = exp[deg$probe_id,]
rownames(exp) = deg$symbol
diff_gene = deg$symbol[deg$change !="stable"]
load("../pipeline/GEO自己练习-GPL6887/lianxi_diff_gene.Rdata")
intersect(diff_gene,diff_gene2)
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
boxplot(exp) #查看exp的阈值
- 1

