
热门标签
热门文章
- 1OpenCV-python图像处理(包含OpenCV库安装)_清华源安装opencv
- 2计网 数据链路层_交换机polling
- 3react18+antd5从0到1的后台管理系统(一)_react18开发管理后台
- 4钉钉在线求饶?爬取钉钉App Store真实评价数据并分析
- 5idea回滚以及码云仓库回滚
- 6左神算法学习日记——单调栈_单调栈 左神模版
- 7浏览器安全_xss 攻击是不是关闭浏览器就会断开
- 8MacOS 下载 brew_brew下载
- 9Python多种方式实现”欢迎小红“_print("你好\nhello") print("你好\\hello") print('小红说:"
- 1014个适合后台管理系统快速开发的前端框架_后台管理框架
当前位置: article > 正文
交互式探索微生物群落与生态功能的关系
作者:知新_RL | 2024-04-27 20:17:27
赞
踩
交互式探索微生物群落与生态功能的关系
微生物群落在生态系统中发挥则重要功能,我们在对微生物群落进行分析时,会将不同分类水平(从门到属)的微生物类群的相对丰度与测定的某一生态功能进行相关性分析。但由于微生物类群数较多,又有不同的分类水平,将其包揽在一个图中,并显示不同的微生物类群的名字,会显得十分杂乱,因此我们尝试使用交互式的可视化来进行探索性分析。下面我们将结合R语言和javascript,来实现这一任务。考虑到微生物生态学方面的同学对JavaScript可能比较陌生,我们也提供了网络工具,为网友在线探索分析微生物各类群与生态功能的关系提供便利。
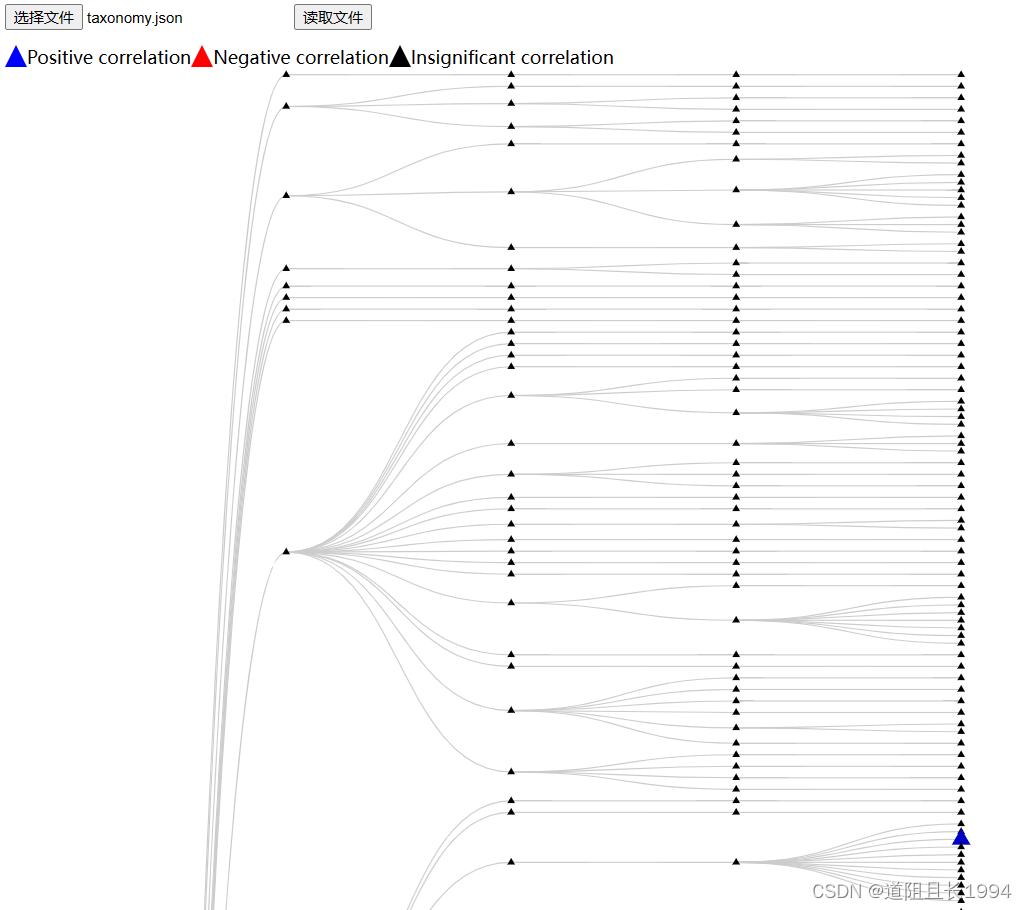
实现的效果:可点击该连接查看实现的效果,TreeCorrelation,鼠标停留在每一个节点可以显示每个节点的微生物类群名称。
例如,当鼠标停留在图中的蓝色节点时会显示其对应的名称,鼠标移开后名称消失,保持图形的整洁。

看完了实现效果,下面具体介绍实现该交互式网页的方法
Step1:数据准备
我们需要使用到两大类的数据:各水平相对丰度表和测定的生态功能表。
- 门水平相对丰度表
- 纲水平相对丰度表
- 目水平相对丰度表
- 科水平相对丰度表
- 属水平相对丰度表
- 生态功能表(测定的生态功能,或者对应的理化性质等)
我们需要使用到个分类水平的相对丰度表,这个在进行完序列分析后都可以获得,其数据格式如下,这里以门水平的相对丰度表为例,纲、目、科、属水平的相对丰度表也类似。
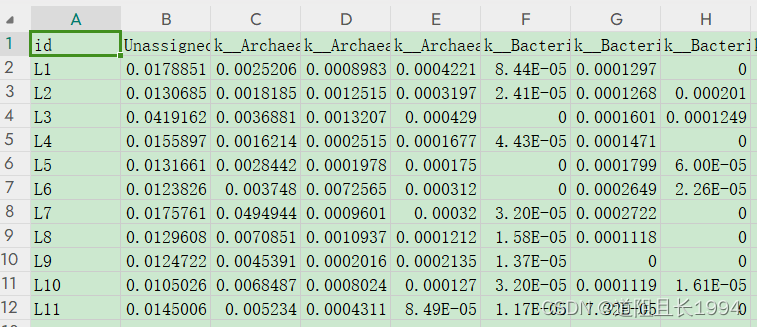
准备好生态功能的数据表
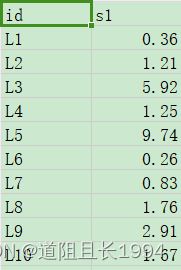
Step2:利用R语言生成JSON文件
2.1 加载必要的包
library(jsonlite)
library(stringr)
library(psych)
- 1
- 2
- 3
2.2 读取文件
L2 <- read.delim("./icicle data/taxa_nifH_16s_L2.txt", row.names=1, check.names = FALSE)
L3 <- read.delim("./icicle data/taxa_nifH_16s_L3.txt", row.names=1, check.names = FALSE)
L4 <- read.delim("./icicle data/taxa_nifH_16s_L4.txt", row.names=1, check.names = FALSE)
L5 <- read.delim("./icicle data/taxa_nifH_16s_L5.txt", row.names=1, check.names = FALSE)
f <- read.delim("./icicle data/FUN.txt", row.names=1)
- 1
- 2
- 3
- 4
- 5
2.3 生态功能与各微生物类群的相关性分析
taxaList <- list(L2=L2,L3=L3,L4=L4,L5=L5)
corList <- list()
for(i in 1:length(taxaList)){
cor <- corr.test(taxaList[[i]],f$s1)
corr <- cor$r
corr[cor$p>0.05] <- 0
corList[[i]] <- corr
}
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
2.4 生成可视化需要的JSON文件
taxa_df <- list() split_string <- strsplit(colnames(taxaList[[length(taxaList)]]), ";") for(i in 1:length(taxaList)){ if(i == length(taxaList)){ taxa_df[[i]] <- colnames(taxaList[[i]]) } else{ taxa_df[[i]] <- sapply(split_string,function(x){paste(x[1:(length(x) - length(taxaList)+i)], collapse = ";")}) } } df <- data.frame(taxa_df) listn <- list() n <- length(df) for(m in 1:n){ list4 <- list() if(m==1){ for (i in 1:nrow(df)){ list4[[i]] <- list(name=df[i,n],value=replace_na(corList[[n-m+1]][i],0)) } listn[[m]] <- list4 } else{ j=1 df0 <- df[!duplicated(df[,n-m+2]),(n-m+1):(n-m+2)] for (i in 1:nrow(df0)){ if (!duplicated(df0[,1])[i]){ list0 <- list(name=df0[i,1],value=replace_na(corList[[n-m+1]][j],0), children=listn[[m-1]][df0[,1] %in% df0[i,1]]) list4[[j]] <- list0 j=j+1 } } listn[[m]] <- list4 } } list1 <- list(list(name="Bacteria",children=listn[[n]]))

- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
2.5 导出JSON文件
json_string <- toJSON(list1, pretty = TRUE)
cat(json_string)
write(json_string,"./taxonomy.json")
- 1
- 2
- 3
Step3:将生成的JSON文件上传网页
打开网页:buildTreeCorrelation,选择并读取生成的JSON文件即可。
声明:本文内容由网友自发贡献,不代表【wpsshop博客】立场,版权归原作者所有,本站不承担相应法律责任。如您发现有侵权的内容,请联系我们。转载请注明出处:https://www.wpsshop.cn/w/知新_RL/article/detail/498512
推荐阅读
相关标签

