
- 1将ElasticSearch的查询结果转为实体类_searchhits转list
- 2苹果 m1 芯片下运行 flink 程序使用 rocksdb 状态后端兼容性问题_flink硬件系统兼容
- 3在做题中学习(54):点名
- 4HT1656 串口配置
- 52024就业热门方向有哪些?这个行业竟然排第一!_优橙教育
- 6公共数据库周报:孟德尔随机化方法
- 7docker部署kafka_docker 部署kafka
- 8rust打包编译为mac或者linux可执行文件,发送到别的电脑不能运行
- 9R数据分析:工具变量回归与孟德尔随机化,实例解析_孟德尔随机化共定位
- 10第四篇【传奇开心果微博系列】Python微项目技术点案例示例:美女颜值判官(1)
甲基化系列 3. 甲基化芯片数据分析完整版(ChAMP)_combat算法
赞
踩

点击关注,桓峰基因
桓峰基因
生物信息分析,SCI文章撰写及生物信息基础知识学习:R语言学习,perl基础编程,linux系统命令,Python遇见更好的你
104篇原创内容
公众号
桓峰基因的教程不但教您怎么使用,还会定期分析一些相关的文章,学会教程只是基础,但是如果把分析结果整合到文章里面才是目的,觉得我们这些教程还不错,并且您按照我们的教程分析出来不错的结果发了文章记得告知我们,并在文章中感谢一下我们哦!
公司英文名称:Kyoho Gene Technology (Beijing) Co.,Ltd.
- 1
如果您觉得这些确实没基础,需要专业的生信人员帮助分析,直接扫码加微信nihaoooo123,我们24小时在线!!
这个ChAMP软件包虽然包括所有的分析内容,但是调用其他包过多,安装时非常麻烦,并且使用了GUI交互界面的功能,个人感觉 体验还是一般吧,建议基础不好的老师还是找桓峰基因,流程已搭建完成,就等待您的创新思想,我们一起合作过,发高分!
前 言
Illumina Infinium HumanMethylationEPIC BeadChip是用于高通量DNA甲基化分析的新平台,与旧的450K阵列相比,有效地增加了一倍的覆盖率。在这里,我们展示了一个显著更新和改进的BioConductor包ChAMP版本,它可以用于分析EPIC和450k数据。添加了许多增强的功能,包括对细胞类型异构性的纠正、网络分析和一系列交互式图形用户界面。

基本原理
ChAMP包设计用于分析Illumina甲基化珠阵列数据(EPIC和450k),并提供了一个集成当前可用的450K和EPIC分析方法的管道。这包括各种不同的数据导入方法(例如从idat文件或beta值矩阵)和质量控制图。Type-2探针校正方法包括SWAN1、Peak Based correction (PBC)和BMIQ(默认选择)。minfi包提供的流行的Functional Normalization函数也是可用的。奇异值分解(SVD)方法允许对批处理效果进行深入研究,为了纠正多个批处理效果,战斗方法已经实现。可以通过RefbaseEWAS对细胞类型的异质性进行调整。ChAMP还包括一个从450k或EPIC数据推断拷贝数变化的函数。对于差异甲基化区域(DMR)的识别,ChAMP提供了新的Probe Lasso方法,以及之前的DMR检测功能Bumphunter和DMRcate。对于需要查找差异甲基化块的用户,ChAMP的新版本包含了检测这些的功能。基因集富集分析(GSEA)也是可能的,新版本的ChAMP包含了一些方法,可以纠正由于基因之间探针的不平等表示而导致的偏差。此外,ChAMP的新版本集成了FEM软件包,可以推断出在用户指定的基因网络中表现出不同表型甲基化的基因模块。虽然有许多用于450k或EPIC数组分析的其他管道和包可用(包括IMA、minfi、methylumi、RnBeads和wateRmelon),但ChAMP提供了一个更全面和完整的分析管道,从读取原始数据文件到最终的第三级分析结果,如GSEA,这为研究人员简化了甲基化阵列分析。新版本ChAMP还提供了一系列基于Shiny和plotly的WebBrower交互分析功能(GUI功能),帮助科学家查看ChAMP的结果。这需要一个基于webbrowser的交互式框架,以便在本地或远程调用Graph System。例如,X11适用于大多数Linux系统。
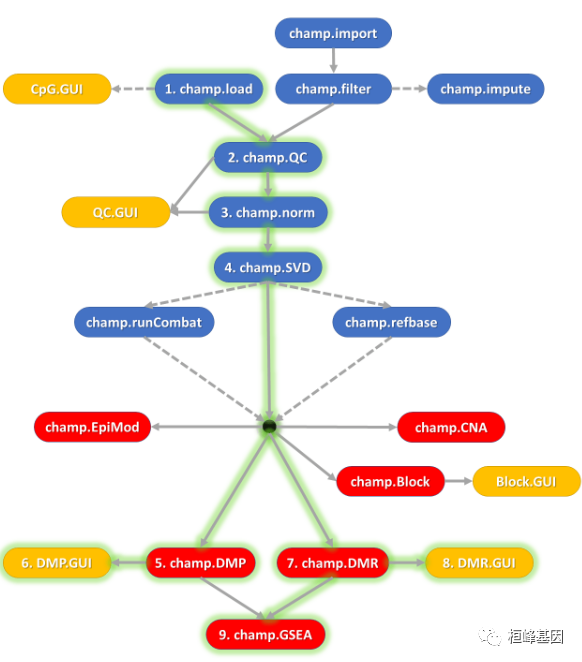
上图概述了ChAMP管道中的步骤。每个步骤都可以作为单独的函数单独运行。这样就可以将ChAMP的结果与其他分析管道进行集成。在上面的管道图中,ChAMP的所有函数都包含在其中,用三种颜色进行分类:蓝色函数表示甲基化数据准备函数,如Loading, Normalization, Quality Control checks等。红色函数表示生成分析结果的函数,如差异甲基化位置(dmp)、差异甲基化区域(DMRs)、差异甲基化块(differential Methylated Blocks)、EpiMod(一种检测来自FEM软件包的差异甲基化基因模块的方法,目前不可用)、通路富集结果等。黄色函数表示数据集和分析结果的GUI界面。上图中的实灰线表示管道流,而虚线表示可能不需要使用的函数,这取决于您的数据和项目。
实例解析
我们两个例子一个是 ChAMP 包自带的测试数据,另外就是我们自己从GEO下载的甲基化数据,大概做到读入即可。
1. 软件安装
当你安装ChAMP时,你可能会遇到一些错误,说有些包没有安装。这些错误是由R包递归导致的,例如,ChAMP使用minfi, minfi使用lumi, lumi使用其他包,所以如果你的电脑上没有安装lumi,ChAMP会失败。要解决这些错误,你只需要检查这些错误信息,找出需要的软件包丢失了,然后直接用BiocManager::install(“YourErrorPackage”)命令安装它。然后重试安装ChAMP,可能需要3-4次,但最终应该可以工作。
if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager")
BiocManager::install(c("minfi", "ChAMPdata", "Illumina450ProbeVariants.db", "sva",
"IlluminaHumanMethylation450kmanifest", "limma", "RPMM", "DNAcopy", "preprocessCore",
"impute", "marray", "wateRmelon", "goseq", "plyr", "GenomicRanges", "RefFreeEWAS",
"qvalue", "isva", "doParallel", "bumphunter", "quadprog", "shiny", "shinythemes",
"plotly", "RColorBrewer", "DMRcate", "dendextend", "IlluminaHumanMethylationEPICmanifest",
"FEM", "matrixStats", "missMethyl", "combinat"))
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
加载软件包,如下:
library(ChAMP) library(minfi) library(Illumina450ProbeVariants.db) library(sva) library(IlluminaHumanMethylation450kmanifest) library(limma) library(RPMM) library(DNAcopy) library(preprocessCore) library(impute) library(marray) library(wateRmelon) library(goseq) library(plyr) library(GenomicRanges)

- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
2. 数据读取
ChAMP包含两个测试数据集,一个是HumanMethylation450数据(.idat),另一个是模拟EPIC数据,可用于测试ChAMP中可用的功能。这可以通过如下所示将目录指向testDataSet来加载
testDir = system.file("extdata", package = "ChAMPdata") myLoad <- champ.load(testDir, arraytype = "450K") ## Failed CpG Fraction. ## C1 0.0013429122 ## C2 0.0022162171 ## C3 0.0003563249 ## C4 0.0002842360 ## T1 0.0003831007 ## T2 0.0011946152 ## T3 0.0014953286 ## T4 0.0015447610 str(myLoad) ## List of 3 ## $ beta : num [1:403116, 1:8] 0.7927 0.6733 0.0794 0.0298 0.0361 ... ## ..- attr(*, "dimnames")=List of 2 ## .. ..$ : chr [1:403116] "cg00000957" "cg00001349" "cg00001583" "cg00002028" ... ## .. ..$ : chr [1:8] "C1" "C2" "C3" "C4" ... ## $ intensity: int [1:403116, 1:8] 2959 8435 8350 6616 4113 3103 15185 7117 9882 2909 ... ## ..- attr(*, "dimnames")=List of 2 ## .. ..$ : chr [1:403116] "cg00000957" "cg00001349" "cg00001583" "cg00002028" ... ## .. ..$ : chr [1:8] "C1" "C2" "C3" "C4" ... ## $ pd :'data.frame': 8 obs. of 8 variables: ## ..$ Sample_Name : chr [1:8] "C1" "C2" "C3" "C4" ... ## ..$ Sample_Plate: logi [1:8] NA NA NA NA NA NA ... ## ..$ Sample_Group: chr [1:8] "C" "C" "C" "C" ... ## ..$ Pool_ID : logi [1:8] NA NA NA NA NA NA ... ## ..$ Project : logi [1:8] NA NA NA NA NA NA ... ## ..$ Sample_Well : chr [1:8] "E09" "G09" "E02" "F02" ... ## ..$ Slide : num [1:8] 7.99e+09 7.99e+09 9.25e+09 9.25e+09 7.77e+09 ... ## ..$ Array : chr [1:8] "R03C02" "R05C02" "R01C01" "R02C01" ...
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
3. 实例操作
在使用这个包时我们可以一次性完成,对自己比较有自信的可以尝试一下,如诺多个地方报错,还是建议分步运行,并且可以清晰的了解每步都在做什么,结果又是怎么解读的?分析之前我们先看下自己电脑的那点资源,是否能满足整套分析,8个CPU,估计够用了,如下:
library("doParallel")
detectCores()
## [1] 8
- 1
- 2
- 3
- 4
一步完成
整个流程可以通过一个命令一次运行,如下:
champ.process(directory = testDir)
- 1
- 2
4. 分步完成
我们注意到champ.process()可能并不总是成功运行,这可能是由于特定数据集的特殊性造成的。例如,pd文件中感兴趣的协变量可以不指定为Sample_Group,而是指定为另一个类别,例如Disease,但是在ChAMP函数中,Sample_Group被默认设置为感兴趣的变量。如果您在使用champ.process()时遇到任何问题,我们建议您循序渐进地应用该软件包。
# Or you may separate about code as champ.import(testDir) + champ.filter() CpG.GUI() champ.QC() # Alternatively: QC.GUI() myNorm <- champ.norm() champ.SVD() # If Batch detected, run champ.runCombat() here. myDMP <- champ.DMP() DMP.GUI() myDMR <- champ.DMR() DMR.GUI() myBlock <- champ.Block() Block.GUI() myGSEA <- champ.GSEA() myCNA <- champ.CNA() # If DataSet is Blood samples, run champ.refbase() here. myRefbase <- champ.refbase()
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
详解步骤
1. 数据过滤
ChAMP提供了一个名为ChAMP .filter()的综合过滤功能,它可以接受任何数据矩阵作为输入(beta、M、Meth、UnMeth、intensity)并对其进行过滤。filter()不调用任何特定的对象或类结果,也不需要IDAT文件,只要有一个beta矩阵,过滤就可以完成。在champ.filter()中,由于一些探针和样本可能会因为质量低而被移除,所以过滤后的数据中可能仍然存在NA。因此,我们在函数中提供了一个参数autoimpute,以便在NAs仍然存在的情况下对剩余探针进行impute。实际上,impute的工作是由champ.impute()函数完成的,读者可以查看该函数以获得更多信息。对于某些过滤方法,必须提供额外的数据,例如,如果希望使用检测p值执行过滤,则需要提供一个检测p值矩阵。同样,如果你想基于beadcount执行过滤,你需要提供beadcount信息。如果使用ChAMP .load()函数的默认方法ChAMP读取IDAT文件,则返回结果中应包含beadcount信息。还champ.filter()的目的是做过滤多种类型的数据帧从相同的数据集,说你有β矩阵,矩阵M,强度矩阵从.idat相同文件,你可以用champ.filter过滤他们()与此同时,这将在未来保持合规分析。首先,必须输入至少一个数据矩阵。其次,如果您提供多个矩阵作为输入,它们必须具有完全相同的行名和名称。例如,如果你想同时对强度数据和M值进行过滤,你必须确保它们有相同的名称,否则,ChAMP会假定它们是来自不同来源的数据帧。然后,champ.filter()将停止过滤进程。由于低质量的样品(如那些有许多未被发现的探测器)可能会在过滤的过程中,重要的是要确保Sample_Name标识符在pd文件一模一样colnames数据矩阵的第四,如果你想做归责,必须提供检测P矩阵,β或M矩阵,且参数ProbeCutoff不能为0,则该参数控制是否应该移除探针的NA比率阈值。如果这三个条件中的任何一个失败,champ.filter()会将autoimpute参数重置为FALSE,则不进行impute处理。第五,如果你想通过珠子数来过滤,你必须提供珠子数,champ.import()会返回珠子信息。或者,你可以使用beadcount函数从西瓜包提取是rgSet对象从minfi方法。ChAMP .filter()函数是ChAMP .import()函数的后继处理,ChAMP .import()函数是ChAMP团队编写的一个新的加载函数。你可以像下面这样使用它们
# Or you may separate about code as champ.import(testDir) + champ.filter() myImport <- champ.import(testDir) myLoad <- champ.filter() head(myLoad$pd) ## Sample_Name Sample_Plate Sample_Group Pool_ID Project Sample_Well Slide ## 1 C1 NA C NA NA E09 7990895118 ## 2 C2 NA C NA NA G09 7990895118 ## 3 C3 NA C NA NA E02 9247377086 ## 4 C4 NA C NA NA F02 9247377086 ## 5 T1 NA T NA NA B09 7766130112 ## 6 T2 NA T NA NA C09 7766130112 ## Array ## 1 R03C02 ## 2 R05C02 ## 3 R01C01 ## 4 R02C01 ## 5 R06C01 ## 6 R01C02 head(myLoad$beta) ## C1 C2 C3 C4 T1 T2 ## cg00000957 0.79274273 0.81628392 0.87269772 0.84761398 0.87328478 0.82818422 ## cg00001349 0.67334505 0.62750070 0.67697354 0.70975670 0.46656322 0.76167976 ## cg00001583 0.07940828 0.07251949 0.37411396 0.15932854 0.44702168 0.14756981 ## cg00002028 0.02977963 0.02320829 0.04002213 0.02730524 0.03504087 0.09662921 ## cg00002719 0.03607880 0.04530982 0.07608838 0.02276532 0.23375170 0.34626219 ## cg00002837 0.21105214 0.27023541 0.34336824 0.43784615 0.13593510 0.41579813 ## T3 T4 ## cg00000957 0.69923664 0.75353389 ## cg00001349 0.24434991 0.45570105 ## cg00001583 0.39635270 0.39144462 ## cg00002028 0.02083333 0.01201803 ## cg00002719 0.07642563 0.30255255 ## cg00002837 0.18168243 0.24598131 head(myLoad$detP) ## NULL
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
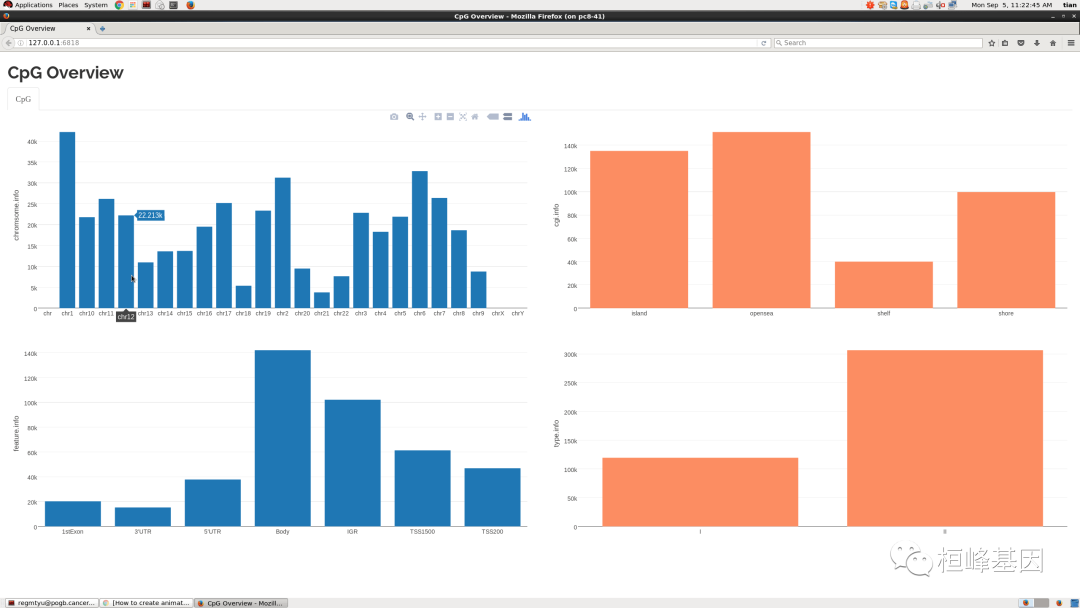
2. CpGs, CpG island, TSS reagions 在基因组上的分布
除了过滤功能,我们还提供了一个CpG.GUI函数,供用户查看自己的CpG在染色体、CpG岛、TSS区域的分布情况。例:这个函数可以用于任何CpG列表,例如,在你的分析过程中,每当你从DMR得到一个重要的CpG列表,你可以使用以下函数来检查你的CpG向量的分布,如下:
CpG.GUI(CpG = rownames(myLoad$beta), arraytype = "450K")
- 1
- 2
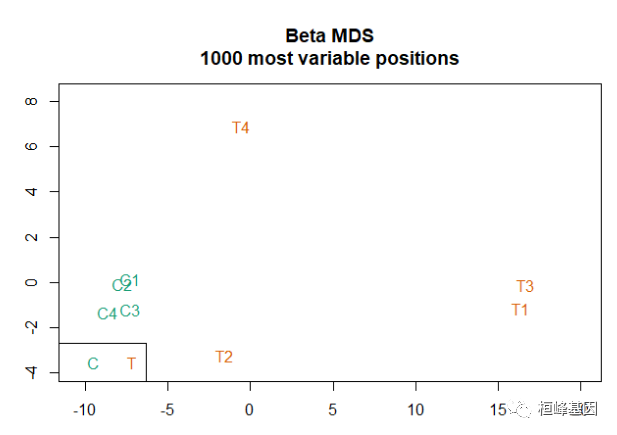
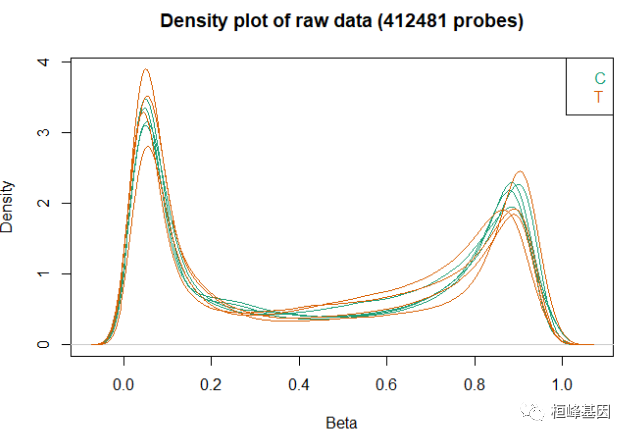
3. 进一步的质量控制和探索性分析
质量控制是保证数据集适合于下游分析的重要环节。champ.QC()函数和QC.GUI()函数会绘制一些图形,方便用户检查数据的质量。通常,会产生三个图,如下:
champ.QC() # Alternatively: QC.GUI()
- 1
- 2
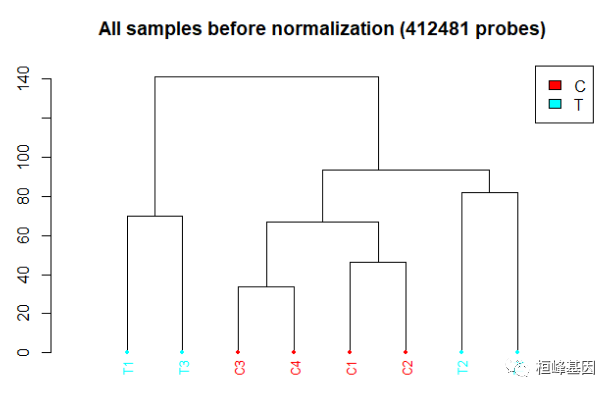
或者你可以使用下面的QC.GUI()函数,但是这可能是内存密集型的,所以当你在ChAMP中运行这个GUI函数时,请确保你有一个好的服务器或计算机。
QC.GUI(beta = myLoad$beta, arraytype = "450K")
- 1
- 2
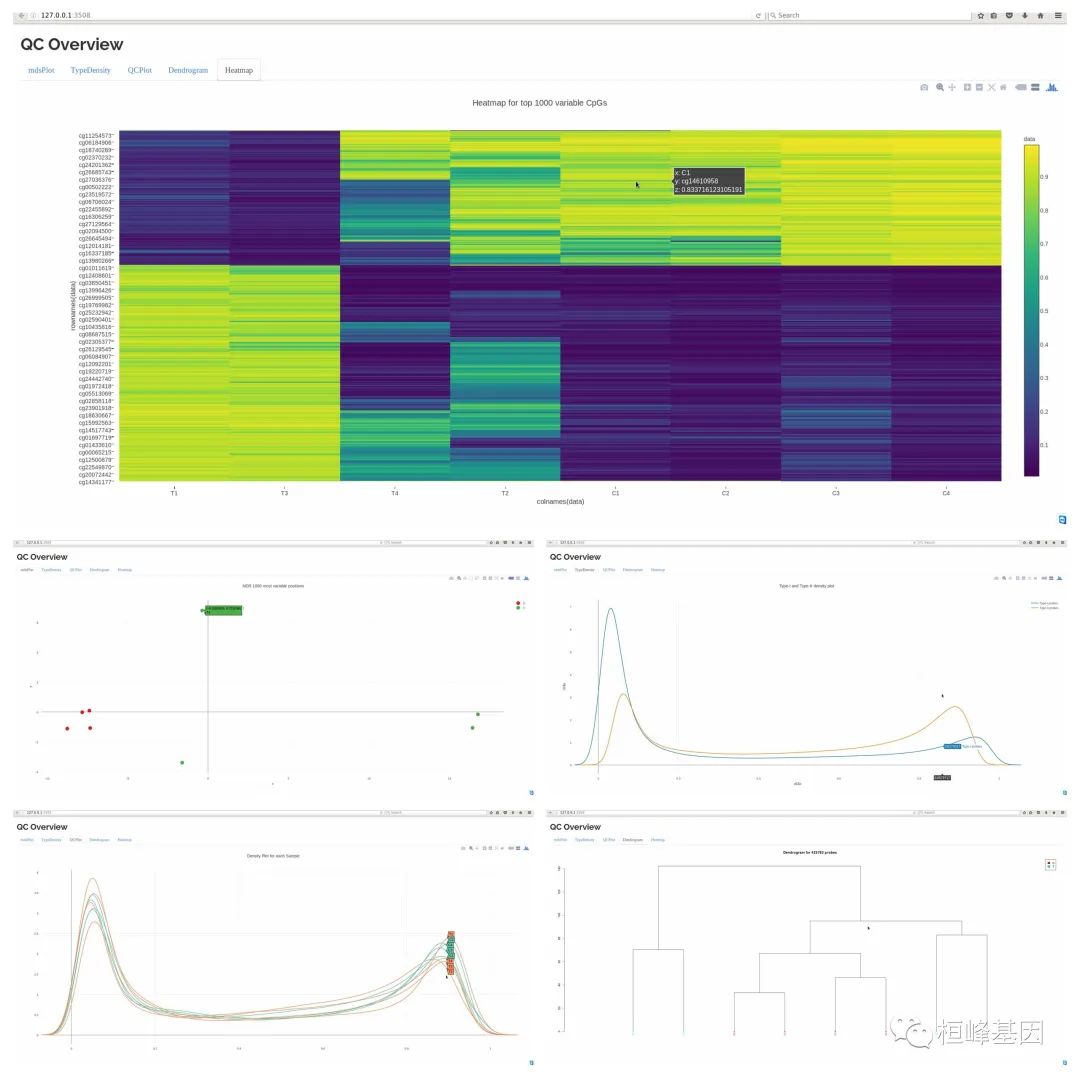
4. 标准化
myNorm <- champ.norm(beta = myLoad$beta, arraytype = "450K", cores = 5)
- 1
- 2
5. SVD 绘制
由Teschendorff7实现的甲基化数据奇异值分解方法(SVD)是评估数据集中显著变异成分数量和性质的强大工具。这些变异的组成部分理想地与生物学因素相关,但通常也与变异的技术来源相关(例如批量效应)。我们强烈建议用户收集尽可能多的被分析样本信息(例如杂交日期、样本收集季节、流行病学信息等),并在与SVD成分相关时将所有这些因素包括在内。如果样品是从.idat文件中加载的,那么如果在champ.SVD()函数中设置参数rgeeffect =TRUE,珠晶芯片上的18个内部探针控件(包括亚硫酸氢盐转换效率)将包括在内。另外,与旧版本的ChAMP包相比,当前版本的ChAMP . svd()会检测所有有效因子来进行分析,这意味着plot包含以下两个条件:协变量至少包含两个值(例如,对于要作为协变量进行测试的BeadChip ID,研究中必须包含来自至少两个不同BeadChip的样本)
champ.SVD(beta = myNorm, pd = myLoad$pd)
- 1
- 2
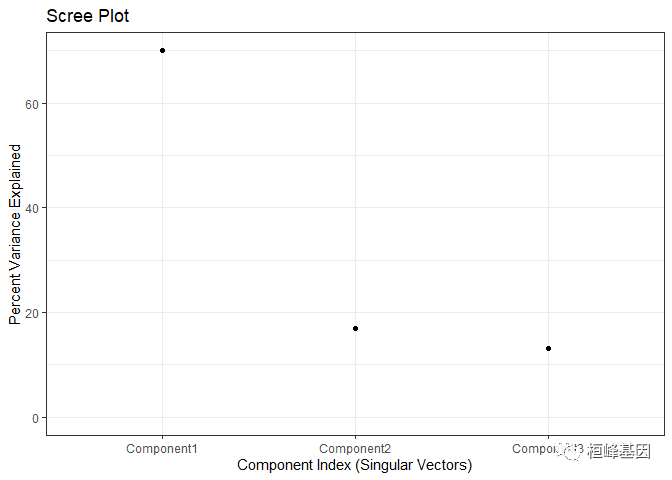
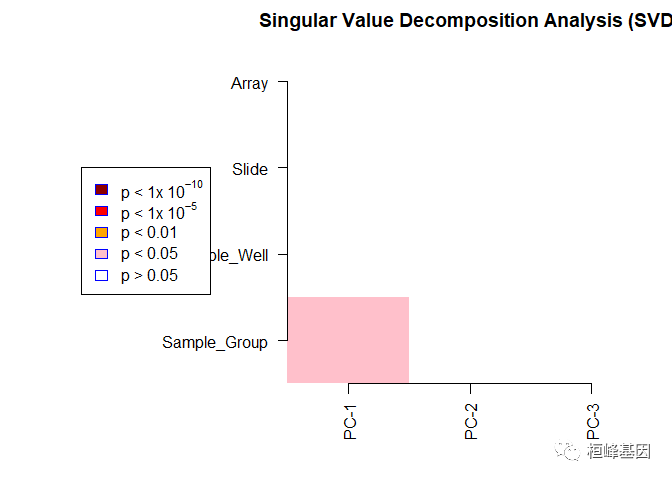
6. 批处理效应校正
ComBat 算法使用经验贝叶斯方法来校正技术偏差。如果ComBat 被直接应用于beta值,输出可能不会被限制在0和1之间。因此,ChAMP logit在ComBat 调整之前转换beta值,然后在ComBat 调整之后计算反向logit转换。如果你有大量的样本,那么ComBat函数将是一个非常耗时的步骤。修正后,可以使用champ.SVD()函数来检查修正后的结果。
myCombat <- champ.runCombat(beta = myNorm, pd = myLoad$pd, batchname = c("Slide"))
## ~Sample_Group
## <environment: 0x000000004bddbbf8>
## Combat can adjust for 1 covariate(s) or covariate level(s)
## ~Sample_Group
## <environment: 0x000000004bddbbf8>
## Standardizing Data across genes
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
7. 差异甲基化探针
差异甲基化探针(DMPs)是最常见的期望输出。这里用户可以使用champ.DMP()函数来计算差异甲基化,也可以使用DMP.GUI()来检查结果。用户可以使用以下代码获取DMP (我们这里仍然使用Demo 450K测试数据):
myDMP <- champ.DMP(beta = myNorm, pheno = myLoad$pd$Sample_Group) ## Contrasts ## Levels pT-pC ## pC -1 ## pT 1 head(myDMP[[1]]) ## logFC AveExpr t P.Value adj.P.Val B ## cg06822689 0.7282395 0.3989916 28.83324 1.529193e-08 0.00630763 7.738472 ## cg04472725 0.6765678 0.3763086 23.79913 5.796498e-08 0.01195473 7.233934 ## cg00841760 0.2273599 0.5564593 21.14488 1.314188e-07 0.01315503 6.858111 ## cg04645886 0.5617829 0.6524946 19.83929 2.040764e-07 0.01315503 6.634181 ## cg20837557 -0.4137773 0.4511552 -19.80416 2.065875e-07 0.01315503 6.627737 ## cg05572370 0.1712660 0.5875608 19.27688 2.488383e-07 0.01315503 6.528199 ## C_AVG T_AVG deltaBeta CHR MAPINFO Strand Type gene ## cg06822689 0.03487186 0.7631114 0.7282395 3 141516291 R II GRK7 ## cg04472725 0.03802467 0.7145925 0.6765678 3 141516232 F II GRK7 ## cg00841760 0.44277935 0.6701393 0.2273599 7 158362966 F II PTPRN2 ## cg04645886 0.37160320 0.9333861 0.5617829 4 1214608 F II CTBP1 ## cg20837557 0.65804379 0.2442665 -0.4137773 6 146864278 F II RAB32 ## cg05572370 0.50192778 0.6731938 0.1712660 11 2323247 F II TSPAN32 ## feature cgi feat.cgi UCSC_CpG_Islands_Name DHS ## cg06822689 Body island Body-island chr3:141516055-141516639 NA ## cg04472725 Body island Body-island chr3:141516055-141516639 NA ## cg00841760 Body island Body-island chr7:158361909-158362970 NA ## cg04645886 Body opensea Body-opensea NA ## cg20837557 TSS1500 shore TSS1500-shore chr6:146864481-146865426 NA ## cg05572370 1stExon opensea 1stExon-opensea TRUE ## Enhancer Phantom Probe_SNPs Probe_SNPs_10 ## cg06822689 TRUE ## cg04472725 TRUE ## cg00841760 NA ## cg04645886 NA ## cg20837557 NA ## cg05572370 NA low-CpG:2279812-2279967 # myDMP is a list now, each data frame is stored as myDMP[[1]], myDMP[[2]], # myDMP[[3]]... DMP.GUI(DMP = myDMP[[1]], beta = myNorm, pheno = myLoad$pd$Sample_Group)
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
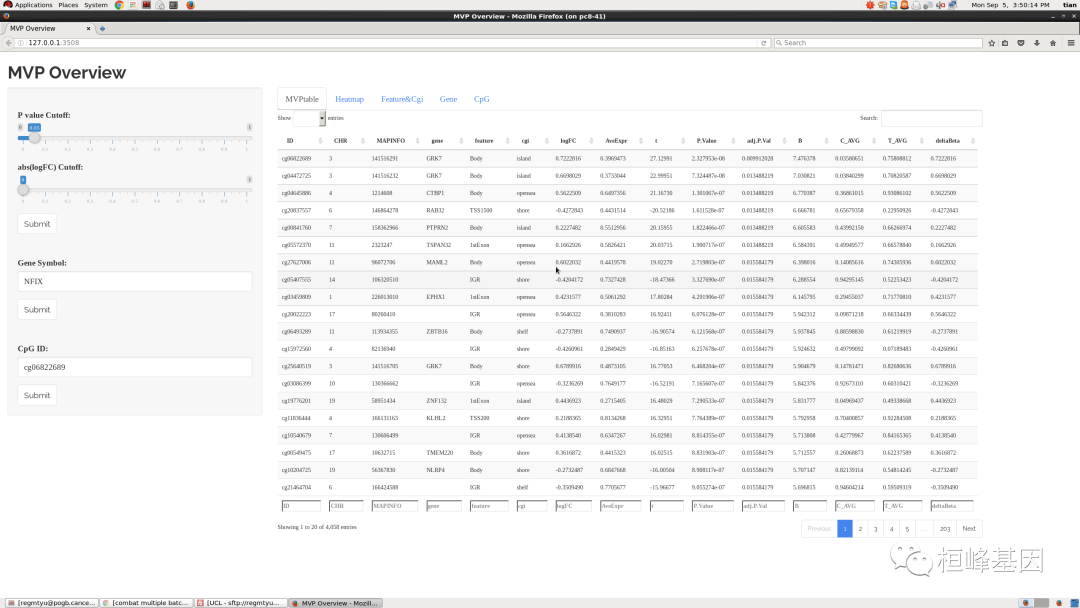
8. 差异甲基化区域
差异甲基化区域(DMRs)是基因组的延伸片段,显示了两组之间DNA甲基化水平的定量变化。ChAMP.DMR()是ChAMP提供的函数,用于计算和返回被划分为离散的差异甲基化区域(DMRs)的探针数据帧,并伴有p值。在ChAMP中实现了三种DMR算法:Bumphunter、ProbeLasso和DMRcate。Bumphunter算法首先将所有探测分组到小簇(或区域),然后应用随机排列法估计候选DMR。这个方法对用户非常友好,并且不依赖于以前函数的任何输出。在Bumphunter中的置换步骤可能需要大量的计算机,但用户可以分配更多的核来加速它。bumphunter算法的结果是一个包含所有检测到的DMR的数据帧,以及它们的长度、簇、注释的CpG数量。
myDMR <- champ.DMR(beta = myNorm, pheno = myLoad$pd$Sample_Group, method = "Bumphunter")
head(myDMR$DMRcateDMR)
## NULL
# It might be a little bit slow to open DMR.GUI() because function need to
# extract annotation for CpGs from DMR. Might take 30 seconds.
DMR.GUI(DMR = myDMR)
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
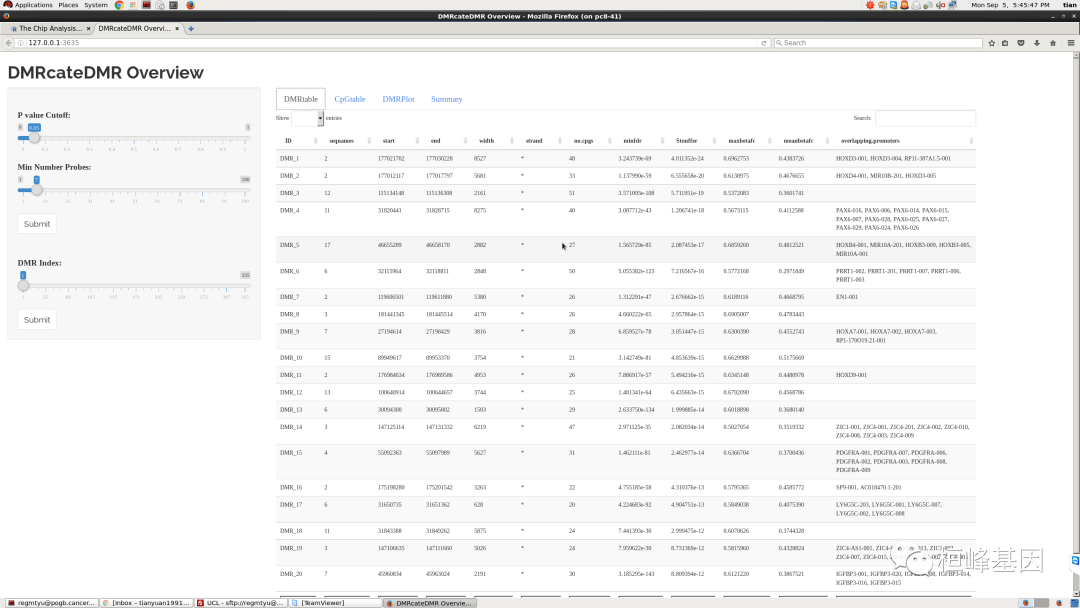
9. 差异甲基化块
识别差异甲基化块(DMBs)也可能是潜在的兴趣。这里我们提供了一个推断DMB的函数。在我们的Block-finder函数中,champ.Block()首先根据它们在整个基因组上的位置计算出小簇(region)。然后对于每个簇(区域),计算其平均值和平均位置,从而可以假设每个区域将被分解为一个单一的单元。当我们发现DMB时,只使用公海上的单个单元进行聚类。这里将使用Bumphunter算法在这些区域上寻找DMRs。在我们之前的论文和其他科学家的工作中,我们证明了差异甲基化块可能在各种癌症中显示普遍特征。近年来,有更多关于差异甲基化块的研究。这里我们提供了一个计算甲基化块的函数。在我们的Block-finder函数中,champ.Block()首先根据它们在整个基因组上的位置计算出小簇(region)。然后对于每个簇(区域),计算其平均值和平均位置,因此可以假设每个区域将被折叠成一个点。然后使用Bumphunter算法在这些区域上寻找DMRs。请注意,只有开放海域将被用于计算区块。差异甲基化块可能在各种癌症中显示普遍特征。我们可以使用以下代码来生成甲基化块:
myBlock <- champ.Block(beta = myNorm, pheno = myLoad$pd$Sample_Group, arraytype = "450K") head(myBlock$Block) ## chr start end value area cluster indexStart indexEnd ## Block_1 11 130933979 133928346 -0.1925868 36.39891 700 21564 21752 ## Block_2 14 100864532 102068637 -0.2215128 35.22054 831 33046 33204 ## Block_3 15 24802619 27360551 -0.1711497 34.40109 838 33611 33811 ## Block_4 7 40026390 42047167 -0.2622415 24.38846 459 84197 84289 ## Block_5 1 247524592 248904369 -0.2476990 18.32972 64 10006 10079 ## Block_6 13 93118732 95086152 -0.2628047 16.55670 790 29120 29182 ## L clusterL p.value fwer p.valueArea fwerArea ## Block_1 189 1558 2.517666e-05 0.042 0.0002781421 0.150 ## Block_2 159 658 2.517666e-05 0.042 0.0003404843 0.184 ## Block_3 201 234 2.517666e-05 0.042 0.0003908376 0.184 ## Block_4 93 651 2.517666e-05 0.042 0.0012947995 0.392 ## Block_5 74 310 5.035331e-05 0.042 0.0030104087 0.510 ## Block_6 63 110 2.517666e-05 0.042 0.0040558394 0.510 Block.GUI(Block = myBlock, beta = myNorm, pheno = myLoad$pd$Sample_Group, runDMP = TRUE, compare.group = NULL, arraytype = "450K")
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
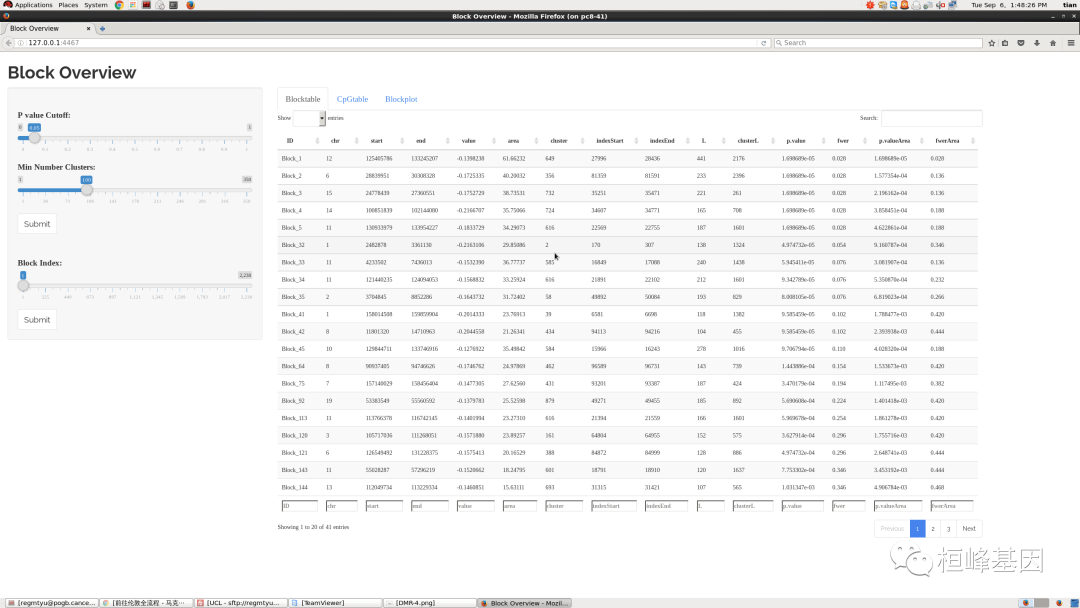
10. 基因集合富集分析
基因集富集分析是大多数生物信息学工作中非常重要的一步。在前面的步骤之后,你可能已经得到了一些重要的DMP或DMRs,因此你可能想知道这些重要的DMP或DMRs所涉及的基因是否在特定的生物学术语或途径中被富集了。要实现这种分析,可以使用champ.GSEA()进行GSEA分析。champ.GSEA()会自动提取myDMP和myDMR中的基因(无论你使用什么方法来生成DMRs)。此外,如果您有自己的显着CpG列表或基因列表,从相同的甲基化阵列(450K或EPIC)计算的其他方法不包括在ChAMP。您也可以将它们格式化为列表,并输入函数来做GSEA(请给您自己的CpG列表或基因列表中的每个元素指定一个名称,否则可能会触发错误)。champ.GSEA()自动提取基因信息,将CpG信息转化为基因信息,然后对每个列表进行GSEA。在CpG到基因的作图过程中,如果有多个CpG被作图到一个基因上,为了避免重复计数,该基因只计算一次。
myGSEA <- champ.GSEA() myGSEA <- champ.GSEA(beta = myNorm, DMP = myDMP[[1]], DMR = myDMR, arraytype = "450K", adjPval = 0.05, method = "fisher") # myDMP and myDMR could (not must) be used directly. head(myGSEA$DMP, 2) ## Gene_List nList nRep fRep ## BENPORATH_ES_WITH_H3K27ME3 BENPORATH_ES_WITH_H3K27ME3 1118 1020 0.9123435 ## TRANSC_FACT TRANSC_FACT 1426 1175 0.8239832 ## nOVLAP OR P.value adjPval ## BENPORATH_ES_WITH_H3K27ME3 228 2.321116 1.311559e-23 1.092266e-19 ## TRANSC_FACT 249 2.172770 2.502087e-22 1.041869e-18 ## Genes ## BENPORATH_ES_WITH_H3K27ME3 NBL1 ASCL1 CALCA DUOXA1 DLX2 PDE4DIP POU3F1 COL27A1 ISL1 PCDHGC4 NEURL SLC6A5 SIX1 KCNIP4 EPHA4 TLE2 GNAO1 SOX17 NEUROG1 HS3ST2 HLA-B KLF4 EPAS1 HOXB13 HLA-F SFRP1 RUNX2 NTRK1 NGFR MAST4 ZIC4 HNF1B EFNA3 HLX CDH6 GPR124 NFIC EXOC3L2 ADAMTS17 ISL2 SPTB BAI2 TNFRSF1B METRNL DUOX1 AGAP2 PHLDB1 CACNA1A HOXA3 POU4F2 DRD4 SIM1 NOXO1 TLX2 ABTB2 PAX1 NTN1 AQP5 ITIH5 LHX5 HOXC12 SLC6A3 HOXB2 HOXC11 CDH23 CACNA1E PLXNA2 WNT10A USH1G ZFHX3 HOXC8 EPHB1 EPB41L4A SCN4B MMP2 PDZD2 RPS6KA2 DLK1 PAX6 LAMB1 FBXL8 TCF21 KCNIP2 EYA4 ADARB2 NFIX CA10 PTGER4 NIN FOXF2 HOXB8 HSF4 TMEM163 SLC1A4 NPAS4 EGFLAM TIGD3 GSN PCDH8 SIX3 HOXD13 HOXB7 ZIC1 KLHL35 KCNQ1 SLCO5A1 GATA3 LGI3 DGKG PRAC SIX2 ANKRD35 ALX4 FOXL2 DLK2 NRG1 ASTN1 CASZ1 HOXD4 PDGFRA PTPRN2 MECOM TBX5 GRIN3A CMTM2 SHOX2 TMEM132E HOXD9 PDE8A PRKCB LHFPL3 NDRG1 WSCD1 CBLN4 NR2E1 FOXD2 CACNG3 FOXA2 EBF1 DPP6 RARA MSX1 IGFBP3 MEOX2 LHX4 WT1 SMOC2 IGSF21 GPR150 DLX1 CCDC140 EBF3 MAB21L1 STX1A OAF CRYL1 HOXB1 PRKCH NRN1 CDH4 CRH B4GALNT1 ZBTB16 CHST8 RAMP1 HHEX SLC35F3 SLITRK3 CYP26C1 FXYD7 MXRA7 HOXD3 KCNJ3 EIF4E3 FLJ45983 CYP24A1 PRRT1 PLXNC1 TBX1 BCAN INSRR CHN2 TP73 HOXB3 NR2F2 CIDEA FOXF1 HOXB6 DRD5 SYNE1 OTOP3 SIX6 HOXA6 CLSTN2 HOXC4 THBS2 HOXA10 SORCS1 HOXC5 SSTR1 ARNTL ST8SIA5 HOXA4 MEIS1 MAN1C1 KCNS2 SOX14 STC2 DLX5 VSX1 HOXA7 LRFN2 DPF3 CCDC50 CYP2A13 HOXC6 NOL4 FLJ32063 ONECUT1 DCLK1 KCNA5 LTBP2 SLC6A2 PKNOX2 LBX1 VAX2 ADAP2 GPR62 ## TRANSC_FACT HIVEP3 RREB1 WT1 ZNF274 NFATC1 MEIS2 HOXC13 ZNF155 SUPT16H SOLH ERG MXD4 LDB2 ZBTB17 ALX4 HHEX DLX5 PBX1 HMGA2 TGFB1I1 TAF2 REPIN1 SMARCAL1 SIX3 NFIC LMO7 JARID2 CBX6 FOXF2 EYA4 SIM1 HOXC4 TOX HOXB9 ZNF574 HOXB5 ARNTL ZNF264 NCOR2 TCF7L1 SP2 NFAT5 PAX1 ESRRB RXRB ONECUT1 HOXA6 ZNF219 MEF2D ZIC4 ZNF550 E2F4 RFX2 HOXD4 SOX14 RUNX2 ZNF154 SHOX2 VSX1 IRF8 MYT1L ASCL1 SOX1 MLLT1 EYA1 PKNOX2 PLAG1 PRDM14 MEOX2 FOXL2 DLX2 ZNF323 TRIM15 ANP32A TFEB CIZ1 MITF NEUROG1 LHX1 GREB1 SOX17 LHX5 ARID3A FOXF1 ZNF212 EP400 HOXB6 TRIM58 EMX2 NR2E1 RORA POU5F1 TBX5 GTF2IRD1 PRRX1 SOX11 HOXB2 TBX1 NFYC MAFF KCNIP2 CSRP1 FOXA2 NR2F2 NFIX PCGF3 TEF POU3F1 ZNF132 LBX1 ZIC1 HOXA5 ISL1 EGLN1 TCF12 TP73 TCF7L2 ZNF395 FOXP1 HOXA10 FOXJ3 BCL11A PACS2 CASZ1 AFF1 FOXD2 HOXB13 MNDA BRF2 SOX12 HOXA3 IRF1 CEBPG PRDM11 RYBP PER3 HOXD3 ZKSCAN1 SIX1 THRB RB1 MEOX1 PURA NOTCH1 HOXC6 MSX1 TCF21 KLF7 HOXD13 HSF4 RING1 HOXB7 POU2AF1 KLF11 NCALD TBX4 TP53BP2 ELF3 HLF EPAS1 GLI3 HOXB3 DPF3 TRIM14 BACH2 TSC22D4 ESRRG RARA KIAA0040 EZH1 HOXC11 TBC1D10B TCF3 MAFK SND1 HOXA11 SRCAP ZIC2 SMAD3 RUNX1 RFPL3 MTA1 EGLN2 TRIP4 SIX2 HDAC4 MEIS1 BCL11B MNT HOXA4 UBP1 RUNX1T1 SOX13 ZBTB7B CBX7 TLX2 TRIM26 ZBTB16 FREQ HOXC5 HOXD10 CHD4 RERE POU3F3 HOXD9 HEY2 SIX6 RORC HOXB8 KLF4 PCSK4 ZIM2 ZNF287 FHL2 ZNF177 CREB5 SMAD7 GFI1 NEUROD6 LYL1 VAX2 BMP4 KCNIP4 CREBBP MYCN GATA3 HIVEP1 JMJD1C POU4F2 WDR45L HOXC8 PAX6 PRDM16 SREBF1 FOXC2 SPEN MYT1 GTF3C1 BRD4 RXRA PBX3 TEAD4 RFX1 SPDEF HOXA7 GLI2 HOXB1 PPARGC1A NR1H3 head(myGSEA$DMR, 2) ## Gene_List nList nRep fRep ## BENPORATH_EED_TARGETS BENPORATH_EED_TARGETS 1062 956 0.9001883 ## BENPORATH_ES_WITH_H3K27ME3 BENPORATH_ES_WITH_H3K27ME3 1118 1020 0.9123435 ## nOVLAP OR P.value adjPval ## BENPORATH_EED_TARGETS 53 8.373755 7.019768e-27 5.846062e-23 ## BENPORATH_ES_WITH_H3K27ME3 52 7.579481 1.197123e-24 4.984821e-21 ## Genes ## BENPORATH_EED_TARGETS HOXC5 HOXD4 TP73 HOXD9 HIST1H3I HOXD3 SIM1 HIST1H4F HOXC6 HSPA1L GBX2 MSX1 PRAC WT1 ALX4 EYA4 HOXA9 FOXF1 HIST1H2BK QRFPR HCG9 HOXD12 HIST1H4L PDGFRA HOXA7 FOXG1 PTGDR HIST1H3C BARHL2 ZIC4 HSPA1A LBX1 IGFBP3 EN1 ZIC1 PAX6 PRDM14 HOXA6 HOXD13 TBX5 LHX8 DLX1 NKX2-3 FLJ32063 PAX9 SIM2 HPSE2 PDX1 HIST1H4I NRN1 VAX1 HOXC4 MAB21L1 ## BENPORATH_ES_WITH_H3K27ME3 GBX2 VAX1 HSPA1A LHX8 PCDHGC4 HLA-F ZIC4 SIM2 RFX4 BARHL2 SIM1 HCG9 NKX2-3 FOXG1 PAX6 EYA4 HPSE2 CCNA1 EN1 HOXD13 ZIC1 PRAC PAX9 PTGDR ALX4 HOXD4 PDGFRA TBX5 PDX1 HOXD12 HOXD9 HSPA1L MSX1 IGFBP3 WT1 GPR150 DLX1 MAB21L1 NRN1 HOXD3 PRRT1 TP73 FOXF1 HOXA6 HOXA9 HOXC4 THBS2 HOXC5 HOXA7 HOXC6 FLJ32063 LBX1
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
结果解读
我们已经简要描述了新ChAMP包中包含的各种管道和函数,欢迎大家指正。
References:
-
Yuan Tian,et.al. ChAMP: updated methylation analysis pipeline for Illumina BeadChips, Bioinformatics, Volume 33, Issue 24, 15 December 2017, Pages 3982–3984.
-
Maksimovic J et a. SWAN: Subset-quantile within array normalization for illumina infinium humanmethylation450 beadchips. Genome Biol. 2012;13(6):R44.
-
Teschendorff AE et al . A beta-mixture quantile normalization method for correcting probe design bias in illumina infinium 450 k dna methylation data. Bioinformatics. 2013;29(2):189-196.
-
Aryee MJ, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363-1369.
-
Fortin JP, Triche T, Hansen K. Preprocessing, normalization and integration of the illumina humanmethylationepic array. bioRxiv. Published online 2016. doi:10.1101/065490.
-
Houseman EA, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13(1):1-16.
-
Feber A, et al. Using high-density dna methylation arrays to profile copy number alterations. Genome Biol. 2014;15(2):R30.

