
- 1微信小程序自定义弹出框组件,模拟wx.showModal_wx.showmodal源码
- 2sd卡卡槽_SD卡面包板插槽DIY图解
- 3Unity基础——List的用法_unity list
- 4Vue中使用require.context()自动引入组件和自动生成路由的方法介绍_vue自动引入组件
- 5STM32的三种Boot模式的差异_system memory启动方式
- 6stm32启动过程、cortex-m3架构、堆栈代码位置、编译汇编链接分析
- 7element-ui el-table 数据变化时表格出现抖动_el-table抖动
- 8java搭建阿里云服务器环境(java环境+mysql+tomcat)和部署 JavaWeb 项目到云服务器(十分详细)_tomcat+mysql的项目迁移到移动云上
- 9ue5.2 数字孪生(11)——Web_UI插件网页通信_ue jsonlibrary-5.2下载
- 10【Java学习路线之JavaWeb】Spring Cloud教程(非常详细)_spring cloud web应用
材料论文阅读/中文记录:Scaling deep learning for materials discovery
赞
踩
Merchant A, Batzner S, Schoenholz S S, et al. Scaling deep learning for materials discovery[J]. Nature, 2023: 1-6.
全文速览
这篇文章主要讲了一种名为GNoME的材料发现框架。该框架利用机器学习和高通量计算方法,通过预测材料的稳定性和性质,加速新材料的发现。文章介绍了GNoME的工作原理和方法,并详细描述了如何使用该框架进行材料发现和验证。文章还提供了一些实验证据来验证GNoME的预测结果的准确性。总的来说,这篇文章展示了GNoME作为一种高效的材料发现工具的潜力,并讨论了其在发现稳定晶体和材料性质预测方面的应用。
核心创新点
本文的核心创新点是使用大规模和多样化的第一原理计算训练GNN(图神经网络),从而实现对无机材料的高效发现。通过与先前工作相比,GNoME模型已经发现了220万个稳定晶体,并为材料科学家提供了以前不可能的建模能力。此外,GNoME模型还展现了在未知化学空间和新的下游任务中的发现能力,包括预测动力学性质。这些创新点为材料发现提供了强大的工具,并加速了材料发现的进程。
重点图文解析

要点:图1展示了GNoME模型的性能随着规模扩大的改进情况。图1e展示了通过对晶体进行随机搜索产生的晶体替代数据进行训练的结构模型在处理超出分布范围的任务时的紧凑性。尽管这些例子通常是高能量的局部极小值,并且与通过结构管道生成的数据相比属于分布范围之外的数据,但我们观察到随着规模的扩大,性能明显改善。这些结果表明,最终的GNoME模型是向社区提供通用能量预测器的重要一步,能够通过深度学习处理多样化的材料结构。

要点:图2展示了以下内容:a. 结构中具有四个以上独特元素的材料数量的增长情况。b. 发现的四元材料的相分离能与Materials Project中的相分离能的比较情况。c. GNoME发现的晶体原型的数量,与Materials Project中的原型数量的比较情况。

要点:图3展示了以下内容:a. 在零样本学习的情况下,预训练的GNoME势能在从AIMD中采样的下游数据上的准确性表现。尽管GNoME势能仅在离子弛豫数据上进行了训练,而没有在分子动力学数据上进行训练,但在零样本学习的情况下,它在来自AIMD模拟的新分布的下游数据上表现出了显著的准确性。d. 零样本学习的GNoME势能与现有通用势能模型在批量系统上的零样本力误差的比较。
摘要
【新兴技术的发展与深度学习运用到更多领域】 新型功能材料实现了从清洁能源到信息处理等技术应用的根本性突破。从微芯片到电池和光伏发电,无机晶体的发现一直受到昂贵的试错方法的瓶颈。与此同时,随着数据和计算的增加,语言、视觉和生物学的深度学习模型展示了新兴的预测能力。在这里,我们展示了大规模训练的图网络可以达到前所未有的泛化水平,【材料发现】 从而将材料发现的效率提高一个数量级。以持续研究中发现的 48,000 个稳定晶体为基础,效率的提高使得能够在当前凸包【convex hull:指的是一组点所形成的最小凸多边形或凸多面体。这个凸多边形或凸多面体包围了这组点,使得这个形状内的任意两点之间的连线都完全位于这个形状内部。在材料科学中,convex hull 经常用于描述材料的晶体结构或者原子排列的几何特征】下方发现 220 万个结构,其中许多结构逃过了人类之前的化学直觉。我们的工作代表了人类已知的稳定材料的数量级扩展。最终凸包上的稳定发现将可用于筛选技术应用,正如我们对层状材料和固体电解质候选物的演示一样。在稳定结构中,有 736 个已通过独立实验实现。数亿次第一原理计算的规模和多样性也解锁了下游应用的建模能力,【下游任务】 特别是带来了高度准确和强大的学习原子间势,可用于凝聚相分子动力学模拟和高保真零样本预测离子电导率。
引言
能量有利的无机晶体的发现在固态化学中具有基础科学和技术意义。几十年来的实验方法已经在无机晶体结构数据库 (ICSD)中编目了 20,000 个计算稳定的结构(总共 200,000 个条目)。然而,由于成本、吞吐量和合成复杂性,这种策略对于规模化来说是不切实际的。相反,材料项目 (MP)、开放量子材料数据库 (OQMD)、AFLOWLIB 和 NOMAD 倡导的计算方法使用基于密度泛函理论 (DFT) 的第一性原理计算作为物理能量的近似值。根据我们自己的重新计算,将从头计算与简单替换相结合,研究人员可以改进到 48,000 种计算稳定的材料(参见方法)。尽管人们一直在寻求有助于进一步发现材料的数据驱动方法,但到目前为止,机器学习技术在估计竞争相能量凸包的稳定性(分解能)方面一直无效。
在本文中,我们通过大规模主动学习扩大了用于材料探索的机器学习,产生了第一个能够准确预测稳定性的模型,从而可以指导材料发现。我们的方法依赖于 【关键两点】 两个支柱:首先,我们建立生成不同候选结构的方法,包括新的对称感知部分替换(SAPS)和随机结构搜索。其次,我们使用最先进的图神经网络(GNN)来改进给定结构或成分的材料属性的建模。 【迭代过程】 一系列的回合中,这些用于材料探索的图网络(GNoME)会被训练使用可用数据来过滤候选结构。经过过滤的候选结构的能量会使用密度泛函理论(DFT)进行计算,这既可以验证模型的预测,也可以作为数据的"飞轮",用于在下一轮主动学习中在更大的数据集上训练更健壮的模型。【数据的飞轮意味着首先使用已有的数据来训练图网络模型(GNoME),然后利用该模型对候选结构进行过滤。接着,通过密度泛函理论(DFT)计算经过过滤的候选结构的能量,这些计算结果又可以被用来验证模型的预测。同时,这些新的计算结果也可以被加入到已有的数据集中,用于训练下一轮的图网络模型。】
通过这个迭代过程,GNoME 模型发现了超过 220 万个相对于之前的工作稳定的结构,特别是包含计算和实验结构的聚合数据集。鉴于已发现的材料竞争稳定性【"已发现的材料竞争稳定性"指的是已经被发现的各种材料之间相互竞争以确定其在特定条件下的稳定性。在材料科学中,稳定性是指材料在特定环境和条件下能够保持其结构和性质的能力。因此,当多种材料在相同或类似的条件下进行比较时,它们会相互竞争以确定哪种材料在给定条件下更为稳定。这种竞争有助于确定最有前景的材料,并为材料设计和开发提供重要的指导】,更新后的凸包由 381,000 个新条目组成,总共 421,000 个稳定晶体,代表了之前所有发现的一个数量级的扩展。与机器学习其他领域的观察结果一致,我们观察到我们的神经网络预测随着数据量的增加呈幂律提高。【结果】 最终的GNoME模型能够准确地预测每个原子的能量到11毫电子伏特,并将基于结构方面进行稳定性预测的精度(命中率)提高到80%以上,仅基于材料的成分信息进行的预测的准确率为33%,而之前的工作中为1% 。此外,这些网络发展出新兴的分布外泛化。例如,GNoME 能够准确预测具有 5 个以上独特元素的结构(尽管在训练中遗漏了),提供了有效探索该化学空间的首批策略之一。我们通过将预测与实验和更高保真度的
r
2
r^2
r2SCAN(参考文献 29)计算进行比较来验证研究结果。
最后,我们证明 GNoME discovery 中生成的数据集为下游应用程序解锁了新的建模功能。结构和松弛轨迹提供了一个庞大且多样化的数据集,可以以前所未有的精度和零样本泛化来训练学习的等变原子间势。我们通过分子动力学模拟估计离子电导率来证明这些潜力对于材料性能预测的前景。
生成和过滤概述
可能材料的空间太大,无法以公正的方式进行采样。由于没有可靠的模型来廉价地近似候选者的能量,【首先】 研究人员通过化学直觉限制生成来指导搜索,通过替换相似离子或枚举原型来完成。尽管提高了搜索效率,但这种策略从根本上限制了候选者的多样性。【进一步】 通过神经网络引导搜索,我们能够使用多样化的方法来生成候选结构,并在不牺牲效率的情况下对晶体空间进行更广泛的探索。
为了生成和过滤候选结构,我们使用两个框架,如图 1a 所示。【第一个】【生成】 首先,通过修改现有晶体来生成候选结构。然而,我们通过调整离子取代概率来大力增强取代集,以优先发现发现,并使用新提出的对称感知部分取代(SAPS)来有效地实现不完全取代。此次扩展导致主动学习过程中超过
1
0
9
10^9
109 候选结构;【过滤】 由此产生的结构通过 GNoME 进行过滤,使用基于体积的测试时间增强和通过深度集成的不确定性量化。最后,对结构进行聚类并对多晶型物进行排序,以使用 DFT 进行评估(参见方法)。 【第二个】 在第二个框架中,成分模型在没有结构信息的情况下预测稳定性。输入是简化的化学式。通过氧化态平衡生成通常过于严格(例如,忽略
L
i
15
S
i
4
Li_{15} Si_{4}
Li15Si4)。使用宽松的约束(参见方法),我们使用 GNoME 过滤组合,并初始化 100 个随机结构,以便通过从头开始随机结构搜索 (AIRSS) 进行评估。
在这两个框架中,模型提供能量预测,并根据竞争相的相对稳定性(分解能量)选择阈值。评估是通过维也纳从头算仿真包 (VASP) 中的 DFT 计算进行的,我们与材料项目相比,测量了发现的稳定材料的数量以及预测的稳定材料的精度(命中率)。
GNoME
所有 GNoME 模型都是预测晶体总能量的 GNN。通过元素的单热嵌入将输入转换为图表。我们遵循消息传递公式,其中聚合投影是具有快速非线性的浅多层感知器(MLP)。对于结构模型,我们发现通过整个数据集中原子的平均邻接度来标准化从边缘到节点的消息非常重要。初始模型是根据 2018 年材料项目的快照进行训练的,其中包含约 69,000 种材料。之前的工作以 28 meV 每原子的平均绝对误差 (MAE) 为基准对这项任务进行了基准测试(参考文献 37);然而,我们发现改进的网络实现了 21 meV每原子的 MAE。我们修复了这个有前景的架构(参见方法),并在本文的其余部分重点关注扩展。
主动学习
我们加速材料发现框架的核心步骤是主动学习。在结构和组成框架中,使用 GNoME 过滤的候选结构是使用 DFT 计算和材料项目中的标准化设置进行评估的。松弛结构产生的能量不仅验证了晶体结构的稳定性,而且还被纳入迭代主动学习工作流程中,作为候选生成的进一步训练数据和结构。尽管结构框架和组合框架的命中率一开始分别低于 6% 和 3%,但通过六轮主动学习,性能稳步提高。 GNoME 模型的最终集成在松弛结构上的预测误差提高到 11 meVatom−1,命中率分别超过 80% 和 33%,清楚地显示了规模的好处。图 1d 提供了最终 GNoME 命中率的分析。
缩放法则和泛化
GNoME 模型的测试损失性能随着进一步的数据呈现出幂律的改进。这些结果符合深度学习中的神经尺度定律28,38,并表明进一步的发现工作可以继续提高泛化能力。需要强调的是,与语言或视觉的情况不同,在材料科学中,我们可以继续生成数据并发现稳定的晶体,这些晶体可以重复使用以继续扩大模型。我们还通过测试结构模型来证明对分布外任务的新兴泛化,这些结构模型是根据图 1e 中随机搜索26产生的晶体替换数据训练的。与我们的结构管道(通过替换,包含接近最小值的结构)生成的数据相比,这些例子通常是高能局部最小值并且不分布。尽管如此,我们观察到规模上的明显改善。这些结果表明,最终的 GNoME 模型是向社区提供通用能量预测器迈出的重要一步,能够通过深度学习处理不同的材料结构。
发现稳定晶体
使用所描述的扩展深度学习材料探索的过程,我们将已知稳定晶体的数量增加了几乎一个数量级。特别是,GNoME 模型发现 220 万个晶体结构对于材料项目来说是稳定的。其中,381,000 个条目作为新发现的材料存在于更新的凸包上。与其他有关结构预测的文献一致,未来的发现可能会将 GNoME 材料从凸包上剔除,类似于 GNoME 取代材料项目和 OQMD 中至少 5,000 种“稳定”材料的方式。有关改进已发现组合物结构的讨论,请参阅补充说明 1。尽管如此,无花果。图 1 和图 2 总结了稳定材料,图 1b 重点关注随时间的增长。我们在图 2a 中看到具有四个以上独特元素的结构数量大幅增加。这是特别有希望的,因为事实证明这些材料对于以前的发现工作来说是困难的27。我们的缩放 GNoME 模型克服了这一障碍,并能够在组合大区域中进行有效发现。通过原型分析进行聚类39支持GNoME发现的晶体的多样性,导致图2c中超过45,500个新原型(比材料项目的8,000个增加了5.6倍),这不可能是通过完全替换或原型枚举产生的。最后,在图2b中,我们将发现的四元系的相分离能(也称为分解焓)与材料项目中的相分离能进行比较,以测量所有其他竞争相到凸包的相对距离。分布上的相似性表明,所发现的材料在竞争相方面具有有意义的稳定性,而不仅仅是“填充凸包”。补充说明 3 中给出了对更新凸包附近(但不在更新凸包上)的材料的进一步分析。
通过实验匹配和 r 2 S C A N r^2SCAN r2SCAN 进行验证
GNoME 的所有候选数据库均源自 2021 年 3 月创建的数据库快照,包括 Materials Project 和 OQMD。在我们进行发现工作的同时,研究人员继续通过实验创造新的晶体,为验证 GNoME 的发现提供了一种方法。在 ICSD 中汇总的实验结构中,有 736 个匹配结构是通过 GNoME 独立获得的。图 1c 中展示了六个实验匹配的结构,补充说明 1 中提供了实验匹配的更多详细信息。同样,自快照以来添加到材料项目的 3,182 种组合物中,有 2,202 种可在 GNoME 数据库中找到,91 种可在 GNoME 数据库中找到。 % 结构匹配。对“新”发现的晶体进行的手动检查支持了这一发现,详细信息参见补充说明 4。
我们还验证预测,以确保基于模型的探索不会过度拟合模拟参数。我们注重功能性的选择。标准投影仪增强波 (PAW)-Perdew-Burke-Ernzerhof (PBE) 势提供了适合大规模发现的速度与精度权衡40,41,但 r2SCAN 函数提供了更准确的元广义梯度近似29,42, 43. 84% 的已发现二元和三元材料也呈现负相分离能量(如图 2d 所示,与材料项目中的 90% 比例相当,但运行规模更大)。 86.8% 的测试四元组在 r2SCAN 凸包上也保持稳定。补充说明 2 进一步分析了 PBE 和 r2SCAN 能量之间的差异。
有趣的组合家族
我们强调了比以前的工作大一个数量级的稳定材料目录的好处。在寻找具有某些理想特性的材料时,研究人员经常过滤此类目录,因为计算稳定性通常与实验可实现性相关。我们对三个应用程序进行类似的分析。首先,层状材料是用于电子和能量存储的有前途的系统44。之前研究的方法45表明,与材料项目相比,大约有 1,000 种层状材料是稳定的,而基于 GNoME 的发现,这一数字增加到大约 52,000 种。同样,通过排除过渡金属或按锂含量等过滤器进行整体筛选方法,我们在 GNoME 发现中发现了 528 种有前景的锂离子导体,比原始研究增加了 25 倍46。最后,Li/Mn 过渡金属氧化物是一个很有前景的家族,可以取代可充电电池中的 LiCoO225,并且与最初的 9 个相比,GNoME 发现了与材料项目相关的额外 15 个候选者。
扩大习得的原子间势
稳定晶体的发现过程还提供了稳定材料之外的数据源。特别是,离子弛豫涉及计算不同材料结构的第一原理能量和力。这生成了一个具有前所未有的多样性和规模的数据集,我们探索该数据集来预训练散装固体的通用机器学习原子间势(MLIP)。 MLIP 已成为一种有前景的工具,可通过学习以第一原理精度计算的参考结构的能量和力来加速材料模拟30,47–49。现有的工作通常根据材料训练模型,数据通常从从头分子动力学 (AIMD) 中采样。这明显限制了它们的普遍适用性和采用,需要昂贵的数据收集并从头开始为每个系统训练新的潜力。通过利用来自不同结构松弛的第一性原理计算的 GNoME 数据集,我们证明了 MLIP 的大规模预训练使模型能够显示出前所未有的零样本精度,并且可用于发现超离子导体,而无需对任何材料进行训练 -具体数据。
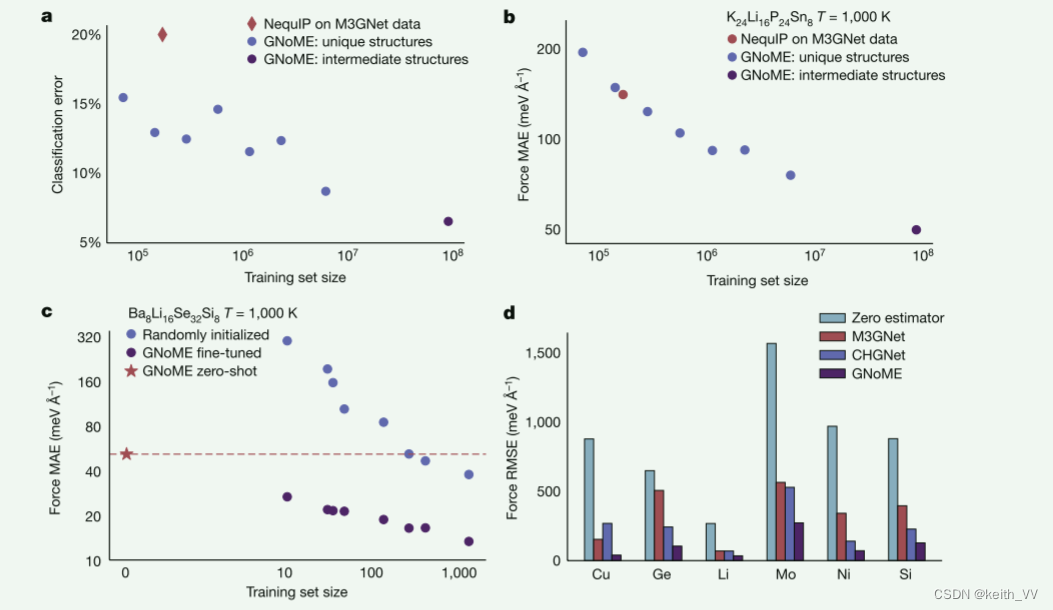
零样本缩放和泛化
我们根据离子弛豫采样的数据对 NequIP 电位 30 进行预训练。增加预训练数据集后,我们观察到准确度呈现一致的幂律改进(见图 3a、b)。尽管只接受了离子弛豫而不是分子动力学数据的训练,但在以零样本方式(即不源自任何训练数据)对从 AIMD 新分布中采样的下游数据进行评估时,预训练的 GNoME 势显示出极高的准确性来自 AIMD 模拟(见图 3)。值得注意的是,这包括看不见的成分、熔化的结构和包括空位的结构,所有这些都不包含在我们的训练集中(参见补充说明 6.4)。特别是,我们发现 GNoME 数据集的规模使其能够超越现有的通用潜力(见图 3d),并使预训练的潜力与在目标数据分布的数百个样本上显式训练的模型竞争(见补充说明) 6.4)。我们观察到 MLIP 的可转让性得到了特别显着的改善,这是 MLIP 最紧迫的缺点之一。为了评估势的可转移性,我们测试了它们在分布偏移下的性能:我们在 T = 400 K 时从 AIMD 采样的结构上训练两种类型的 NequIP 势,一种是根据随机初始化的权重来训练网络,另一种是用随机初始化的权重来训练网络。我们从预训练的 GNoME 检查点进行微调。然后,我们在 T = 1,000 K 时测量从 AIMD 采样的数据上的两种势的性能(见图 3c),相对于 400-K 数据不分布。即使在超过 1,000 个结构上进行训练,在 GNoME 数据上进行预训练的电位也显示出比从头开始训练的电位在可移植性方面有系统且强大的改进。零样本 GNoME 潜力,未针对该组合中的任何数据进行微调,甚至优于在数百个结构上训练的最先进的 NequIP 模型。
筛选固态离子导体
固体电解质是固态电池的核心组成部分,比液体电解质具有更高的能量密度和安全性,但目前离子电导率较低。在寻找新型电解质材料时,AIMD 可以根据第一原理预测离子电导率。然而,由于 DFT 与电子数量的缩放比例较差,常规模拟仅限于数百皮秒、数百个原子,最重要的是,较小的组成搜索空间。在这里,我们表明,GNOME 势在这种分布外、零射击设置中表现出高鲁棒性,并推广到高温,这使得它们能够作为新型固态电解质高通量发现的工具。我们使用在规模不断增大的数据集上预训练的 GNoME 势,对 623 种前所未见的成分进行分子动力学模拟。图 3a 显示了与 AIMD 相比,预训练的 GNoME 势将看不见的成分分类为超离子导体的能力。
当扩展到比现有方法大得多的 GNoME 数据集时,我们发现深度学习解锁了以前不可能的能力,为无机块状晶体构建可转移的原子间势,并允许大规模地对材料特性进行高精度、零样本预测。
结论
我们证明,经过大量多样的第一原理计算训练的 GNN 可以实现无机材料的有效发现,将稳定晶体的数量增加一个数量级以上。相关数据集支持机器学习的原子间势,对不可见的散装材料提供准确而强大的分子动力学模拟。我们的研究结果提出了关于深度学习系统在自然科学中的能力的有趣问题:机器学习方法在科学发现中的应用传统上遇到了基本挑战,即学习算法在训练和测试时数据分布相同的假设下工作次,但发现本质上是一种非分发的努力。我们在大规模学习方面的结果通过证明 GNoME 模型展现出大规模的新兴分布能力,为克服这一困境提供了潜在的一步。这包括在看不见的化学空间(例如,具有四种以上不同元素)中的发现,以及新的下游任务(例如,预测动力学性质)。
相对于之前的工作,GNoME 模型已经发现了 220 万个稳定晶体,并为材料科学家提供了以前不可能实现的建模能力。对于研究成果在应用中的转变,仍然存在一些悬而未决的问题,包括通过竞争多晶型物更好地理解相变、振动曲线和构型熵产生的动态稳定性,以及最终的可合成性。尽管如此,我们看到预训练的通用 GNoME 模型被用作各种应用程序中的强大工具,从根本上加速材料发现。
在线内容
任何方法、附加参考文献、自然组合报告摘要、源数据、扩展数据、补充信息、致谢、同行评审信息;作者贡献和竞争利益的详细信息;数据和代码可用性声明可在 https://doi.org/10.1038/s41586-023-06735-9 上获取。

